Т6.Экстракция в аналитической биохимии и при выделении ксенобиотиков
| Сайт: | Электронный информационно- образовательный портал ВолгГМУ |
| Курс: | Дисциплина Химико-токсикологические исследования в работе клинической лаборатории |
| Книга: | Т6.Экстракция в аналитической биохимии и при выделении ксенобиотиков |
| Напечатано:: | Гость |
| Дата: | Четверг, 9 Май 2024, 22:00 |
Оглавление
- 1. ЭКСТРАКЦИЯ КАК МЕТОД РАЗДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ
- 2. ЧТО ТАКОЕ ЭКСТРАКЦИЯ
- 3. ЭКСТРАКЦИЯ КАК ХИМИЧЕСКАЯ РЕАКЦИЯ. Основные законы
- 4. КЛАССИФИКАЦИЯ ЭКСТРАКЦИОННЫХ ПРОЦЕССОВ
- 5. Экстракция в органической химии
- 6. Пробоподготовка в аналитической токикологии
- 7. Выделение ДНК из лейкоцитов крови
- 8. Выделение ДНК из крови с помощью смолы Chelex
- 9. Экстракция липидов
1. ЭКСТРАКЦИЯ КАК МЕТОД РАЗДЕЛЕНИЯ И КОНЦЕНТРИРОВАНИЯ
Успешное решение проблемы охраны биосферы, снижение отрицательного влияния индустриализации на состояние природной среды и многие другие глобальные проблемы непосредственно связаны с разработкой эффективных методов анализа. Состояние методов избирательного определения металлов не всегда удовлетворяет требованиям к нижним границам определяемых содержаний. Постоянно ощущается необходимость в простых по выполнению, точных, чувствительных методиках, которые позволяли бы определять компонент в сложной по составу смеси. Для решения этой проблемы ученые привлекли методы концентрирования, которые позволили в значительной степени устранить сложные ситуации. Более того, в некоторых случаях концентрирование расширило пределы применимости инструментальных методов (атомно-абсорбционной спектрометрии, хроматографии, спектрофотометрии, вольтамперометрии).
Одним из перспективных методов разделения и концентрирования является экстракция [1]. Давно известно, что многие вещества распределяются между двумя несмешивающимися жидкостями, причем характер разделения в известной степени зависит от растворимости веществ в индивидуальных фазах. Использование этого явления для разделения и очистки веществ стало одним из основных методов в органической химии. Долгое время в области неорганической химии экстракцией интересовались мало. Поэтому экстракцию принято считать относительно молодым методом. Это в значительной мере оправданно, особенно если принять во внимание, что наиболее мощный толчок развитие экстракции получило только в середине нынешнего столетия в связи с работами в области ядерной технологии. Работы в области экстракции микроэлементов проводились, конечно, и ранее. Элементный бром экстрагировали еще в 1825 году. В 60-е годы прошлого века была предложена экстракция роданидов металлов. Этот метод применяется до сих пор. В 20-е годы Фишер исследовал экстракцию комплексов металлов с дитизоном и выявил зависимость распределения элементов от концентрации реагента, металла и ионов водорода.
Начало количественному описанию экстракции (с химических позиций) положили Кольтгоф и Сендел, которые вывели в 1941 году уравнение, характеризующее экстракцию хелатов. Ирвинг и Уильямс развили эту теорию. Последующие интенсивные исследования привели к выяснению химизма большинства экстракционных процессов [2]. Современные экстракционные методы достаточно универсальны. Трудно найти типы соединений, которые нельзя было бы экстрагировать. С помощью экстракции можно разделять многокомпонентные системы, причем эффективнее и быстрее, чем это достигается другими методами. Экстракционные методы пригодны для абсолютного и относительного концентрирования, извлечения в экстракт микроэлементов или матрицы, индивидуального и группового выделения элементов. В статье рассматривается современное состояние экстракции микроэлементов и не затрагиваются вопросы экстракции органических соединений.
2. ЧТО ТАКОЕ ЭКСТРАКЦИЯ
Экстракция - это процесс распределения вещества между двумя несмешивающимися растворителями. Одним из них обычно является вода, вторым - органический растворитель. Будучи гетерогенным процессом, экстракция подчиняется правилу фаз Гиббса: N + F = K + 2, где N - число фаз, F - число степеней свободы, K - число компонентов. При экстракции обычно две фазы (N = 2), одно распределяемое вещество (K = 1). Следовательно, при постоянных температуре и давлении система моновариантна (F = 1). Таким образом, если концентрация растворенного вещества в одной фазе постоянна, то его концентрация в другой фазе также постоянна. Соотношение между концентрациями растворенного вещества в каждой из фаз привело к формированию закона распределения.
Выполнение экстракционного разделения и концентрирования обычно не требует сложного и дорогостоящего оборудования. В лаборатории это чаще всего делительная воронка (рис. 1). С помощью воронки проводят так называемую периодическую экстракцию. Обычно водный раствор пробы и органический растворитель тщательно перемешивают встряхиванием вручную или с помощью механического устройства. После разделения фаз нижнюю фазу сливают через кран. Сильное встряхивание нежелательно, так как оно может привести к образованию эмульсий, что затрудняет разделение двух фаз. Если увеличение нужного компонента неполное, экстракцию повторяют, разделив фазы и прибавив к водной фазе новую порцию органического растворителя.
Экстракция - сложный физико-химический процесс. Теория экстракции находится на стыке различных разделов химии: химической термодинамики, теории растворов, химической кинетики, органической химии и координационной химии. Для описания экстракционных процессов необходимо также использовать теорию массопереноса. Задача экстракции состоит в том, чтобы полно и селективно перевести компонент из водной фазы в органическую. Для этого необходимо подобрать условия образования подходящих соединений (например, комплексов металлов), в виде которых компонент может находиться в органической фазе [3].
УСЛОВИЯ ЭКСТРАКЦИИ ВЕЩЕСТВА
Извлечение металла в органическую фазу возможно только в том случае, если растворимость соединений этого металла в органическом растворителе выше, чем в воде. В реальных системах металл существует в виде разнообразных соединений. Следует учитывать, что в ходе экстракции могут образоваться формы, которых в исходном растворе не было. Поэтому прежде всего необходимо установить, в виде какого соединения экстрагируется металл и какова его растворимость. Растворимость любого соединения зависит от многих факторов: природы вещества, температуры и давления. Обычно химически подобные вещества лучше растворяются друг в друге, чем в веществах другой структуры. При этом подобие не следует понимать слишком узко, так как часто уже достаточно присутствия в молекулах одинаковых или сходных по поведению групп. Молекулы растворителя вступают с растворенными молекулами в энергетическое взаимодействие, в первую очередь типа электростатического, так как молекулы большинства растворителей обладают электрическими дипольными моментами.
Одним из условий проведения экстракции является нейтрализация заряда. Заряженные соединения не могут переходить в органический растворитель. Присутствующие в растворе ионы металла необходимо перевести в незаряженный комплекс либо в ионный ассоциат с подходящим ионом противоположного заряда. Величина заряда иона играет существенную роль при экстракции ионных ассоциатов. В этом случае лучше всего извлекаются в органическую фазу однозарядные ионы, хуже - двух- и особенно трехзарядные. Кроме того, экстрагирующееся соединение должно быть гидрофобным и не содержать гидрофильных групп, например гидроксильных или карбоксильных.
3. ЭКСТРАКЦИЯ КАК ХИМИЧЕСКАЯ РЕАКЦИЯ. Основные законы
Процесс экстракции почти всегда можно описывать как обычную, хотя и двухфазную, химическую реакцию. Реакции экстракции практически всегда обратимы, поэтому к экстракционным процессам можно приложить закон действия масс. Появляется возможность говорить о константе равновесия реакции экстракции, которую в данном случае называют константой экстракции. Процесс экстракции металлов можно представить в следующем виде:
Индексы обозначают органическую (о) и водную (в) фазы. Органический реагент (HA) растворим в органическом растворителе. Концентрационная константа равновесия этой реакции (константа экстракции - Kех)
Записывать выражение для константы экстракции можно только в том случае, когда известны состав экстрагирующегося соединения исходных компонентов. Наряду с законом действия масс к экстракционным системам применим, как уже отмечалось, закон распределения, согласно которому при постоянной температуре и давлении отношение равновесных концентраций вещества в двух несмешивающихся фазах является постоянной величиной, не зависящей от общей концентрации вещества. Эта величина называется константой распределения KD :
где [A]о и [A]в - равновесные концентрации вещества в органической и водной фазах. Однако обычно экстрагируемое вещество присутствует в разных формах, особенно при экстракции металлов. Соединения металла участвуют в различных химических превращениях комплексообразования, гидролиза, полимеризации, диссоциации и т.д. В связи с этим отношение общих концентраций металла в органической и водной фазах не является константой. Для каждой отдельной формы металла (Mi) закон распределения должен выполняться, то есть [Mi]о / [Mi]в = KD . Из этого следует, что необходимы количественные характеристики, которые можно было бы измерить в прямом эксперименте. Это прежде всего коэффициент распределения D, представляющий собой отношение общей концентрации вещества в органической фазе к общей концентрации его в воде:
Рассчитать величину D можно определив любым подходящим методом концентрации Cо и Cв . При экстракции металлов используют для этого методы атомно-эмиссионной и атомно-абсорбционной спектрометрии, вольтамперометрию, спектрофотометрию, метод радиоактивных индикаторов [1, 4]. В отличие от константы распределения в случае коэффициента распределения нет требования постоянства и равенства форм существования вещества в общих фазах и требования равновесности системы.
Для внутрикомплексного соединения MAm [4] коэффициент распределения
Коэффициент распределения описывает способность вещества экстрагироваться, но не определяет реальную полноту извлечения, которая зависит от соотношения объемов органической и водной фаз. При одном и том же коэффициенте распределения вещество извлекается тем полнее, чем больше объем органической фазы (при постоянном объеме водной). Долю проэкстрагированного вещества выражают величиной степени извлечения:
где Cв и Cо - количество вещества в органической и водной фазах. Степень извлечения чаще всего выражают в процентах:
где Vо и Vв - объемы органической и водной фаз. В случае равенства объемов фаз (Vо = Vв) получаем
Величину, характеризующую возможность разделения двух веществ, называют коэффициентом разделения (KА/В):
Для хорошего разделения недостаточно только того, чтобы коэффициент разделения был высоким. Необходимо также, чтобы произведение коэффициентов их распределения было близко к единице. В практике используют также коэффициент концентрирования (SА/В): SА/В = RA / RB .
К числу важных факторов, влияющих на экстракцию, относится время контакта фаз. Практическая важность вопроса связана прежде всего с тем, что во многих экстракционных системах равновесие достигается не сразу. Скорость экстракции зависит от скорости химических реакций, протекающих в системе, в частности от скорости массопереноса вещества между двумя фазами. При этом для ускорения экстракции необходимо использовать различные факторы. Если наиболее медленным является массоперенос, следует, например, увеличить скорость перемешивания фаз. На скорость химических реакций можно влиять увеличивая концентрации взаимодействующих компонентов, подавляя мешающие реакции, например гидролиза и полимеризации [3]. Экстракционное равновесие быстро достигается при извлечении ионных ассоциатов, когда при экстракции не меняется внутренняя координационная сфера центрального атома металла, а происходит лишь взаимодействие ионов. Наиболее медленно экстракция протекает в случае металлов, образующих кинетически инертные комплексы, например хрома (III) или некоторых платиновых металлов.
Таким образом, на практике при разработке экстракционных методов необходимо изучать скорость реакции. Для этого следует установить время достижения равновесия. Обычно это делают по кинетическим кривым, например, зависимости коэффициента распределения от времени контакта фаз (рис. 2). Изучение зависимости степени извлечения от времени контакта фаз может не дать правильной информации об установлении равновесия, если коэффициенты распределения достаточно высоки, что видно из рис. 3. Хотя коэффициенты распределения продолжают расти, то есть равновесия нет, степень извлечения составляет 100% и может создаться впечатление, что система находится в равновесии.
Одним из интересных в теории экстракции является вопрос о том, где образуется экстрагирующееся соединение - в водной фазе, в органической или на границе их раздела. Общего решения, по-видимому, быть не может. В разных системах это может быть по-разному, но изучение скорости экстракции, например различными растворителями, позволяет решить этот вопрос. Одним из способов решения может служить изучение кинетики экстракции внутрикомплексных соединений различными растворителями. Растворители нужно выбирать таким образом, чтобы константы распределения реагента (КD,НА) в них различались. Условия проведения экстракции должны быть такими, чтобы определяющей в кинетическом отношении была скорость химического взаимодействия. При этом рН, концентрация реагента должны быть одинаковы. Если соединение образуется в водной фазе, скорость экстракции будет тем больше, чем выше равновесная концентрация органического реагента в водной фазе. Чем ниже КD,НА , тем больше концентрация. Иными словами, чем хуже растворим экстрагент в органическом растворителе, тем больше скорость экстракции. Если соединение образуется на границе, то, чем лучше растворим реагент в органическом растворителе, тем больше будет скорость экстракции. Образование соединения в органической фазе вряд ли возможно, так как для этого экстрагируемый элемент должен каким-то другим путем перейти в органический растворитель. Необходимо отметить, что проблема эта сложная и методы ее решения, рассмотренные выше, не являются идеальными.
4. КЛАССИФИКАЦИЯ ЭКСТРАКЦИОННЫХ ПРОЦЕССОВ
Экстракционные системы весьма разнообразны. Рациональный подбор системы в значительной мере определяет успех экстракционного концентрирования. Поэтому классификация экстракционных процессов и экстрагирующихся соединений имеет важное значение. Положив в основу классификации характер соединения, переходящего в органическую фазу, можно выделить два основных типа: неионизированные соединения и ионные ассоциаты. Более тонкая классификация позволяет выделить несколько групп соединений, относящихся к тому или иному типу.
Внутрикомплексные соединения (ВКС) - один из самых распространенных классов соединений, используемых при концентрировании микроэлементов [3], поэтому рассмотрим эту группу соединений подробнее. Экстракцию ВКС широко используют в аналитической химии, радиохимии, цветной металлургии. Реагент, образующий ВКС, должен содержать по крайней мере два атома, способных одновременно координироваться металлом (например, O, S, N). Одна из активных групп в молекуле реагента должна включать подвижный атом водорода (HA), замещаемый в процессе комплексообразования на металл. Вторая группа может быть также кислотной или основной. Применяют реагенты, являющиеся слабыми кислотами. Число кислотных групп может быть различным, но обычно используют многоосновные кислоты, являющиеся бидентатными лигандами. Полидентатные реагенты более избирательны.
Таким образом, по формуле реагента можно определить, способен ли он образовывать ВКС. Иногда это трудно сделать, так как реагент подвергается превращениям (таутомерия), которые изменяют его структуру и комплексообразующие свойства. Например, в случае b-дикетонов:
Комплексы с металлами образует форма II. Таутомерные превращения наблюдаются и в случае дитизона:
Обратимся к примерам ВКС, образуемых бидентатными реагентами. Экстракцию диэтилдитиокарбамата меди (V), диметилглиоксимата никеля (VI), дитизоната свинца (IV), оксихинолината цинка используют в аналитических лабораториях всего мира:
Для ВКС характерны высокие коэффициенты распределения несмотря на невысокую растворимость комплексов в органических фазах. Из-за малой растворимости емкость экстрактов не очень велика, но в некоторых случаях вполне достаточна даже для технологических целей. ВКС часто окрашены, что обеспечивает их использование для фотометрического определения элементов. Некоторые ВКС термически устойчивы и летучи, что дает возможность сочетать экстракцию с газохроматографическим разделением и определением металлов.
Остановимся на количественных характеристиках процесса экстракции ВКС. В условиях, когда в водной фазе можно пренебречь всеми формами металла, кроме иона Mn+, отношение MAn(0)/ Mn+ выражает коэффициент распределения D. С учетом этого преобразуем выражение (2):
Это основное уравнение, описывающее экстракцию ВКС. В логарифмической форме:
lg D = lg Kex + n lg [HA]о + npH.
При постоянной равновесной концентрации реагента в органической фазе имеем линейную зависимость логарифма коэффициента распределения от pH с наклоном, равным n. Увеличение pH на единицу повышает коэффициент распределения в 10 раз для однозарядного иона металла, в 100 раз для двухзарядного, в 1000 раз для трехзарядного и т.д. [3]. Типичная кривая, характеризующая зависимость экстракции ВКС от pH, выглядит так, как показано на рис. 4. По мере повышения pH в водной фазе, кроме иона Mn+, начинают появляться низшие неэкстрагируемые комплексы с реагентом. Поэтому зависимость начинает отклоняться от прямой и в той области, где в обеих фазах существует MAn , выходит на плато (рис. 4). Нисходящая ветвь характеризуется существованием в водной фазе анионных комплексов. Уменьшение экстракции может быть вызвано также гидролизом. Зависимость экстракции от pH при использовании данного реагента часто неодинакова для различных элементов. Это имеет большое значение, так как позволяет, регулируя pH, осуществлять эффективное разделение. Экстрагируя ионы металлов в виде их диэтилдитиофосфатов, можно путем изменения кислотности среды выделить группы элементов, различающиеся по их электрохимической активности [5].
Константа экстракции зависит также от ряда констант константы устойчивости экстрагирующегося комплекса bn , константы диссоциации реагента KHA , константы распределения комплексов KD, MA и константы распределения реагента KD, HA . Зависимость эта выглядит следующим образом:
Таким образом, экстракция тем лучше, чем выше устойчивость комплекса и чем больше его константа распределения. Далее экстракция тем выше, чем более сильной кислотой является реагент и чем меньше он переходит в органическую фазу.
КАК ВЫБРАТЬ ЭКСТРАГЕНТ
При экстракции ВКС выбор реагента основан главным образом на знании указанных выше констант, соотношении их величин. Чем выше KНА , тем ниже bn , то есть с точки зрения влияния на константу экстракции эти величины действуют в разных направлениях. То же и с константами распределения (KD, M и KD, HA). Поэтому в каждом случае надо решать задачи оптимизации.
Экстрагенты для экстракции координационно сольватированных нейтральных комплексов удобно подбирать руководствуясь принципом жестких и мягких кислот и оснований (ЖМКО) [6]. Подобные комплексы экстрагируются только при использовании экстрагентов, способных к координации с металлом. В большинстве случаев это электронодонорные экстрагенты, имеющие свободную пару электронов. В первую очередь нужно назвать кислород, азот и серосодержащие экстрагенты. При образовании смешанных комплексов рассматриваемой группы металл почти всегда выступает в роли акцептора электронов [2].
Для жестких по ЖМКО редкоземельных элементов, актинидов, циркония, железа подходят жесткие кислородсодержащие нейтральные экстрагенты. Для мягких платиновых металлов, ртути, кадмия, висмута, серебра лучше серосодержащие. Для промежуточных переходных dэлементов, таких, как медь или никель, - и те и другие. Если экстрагент наряду со способностью входить во внутреннюю координационную сферу металла к тому же легко протонируется, то в принципе наряду с нейтральными смешанными комплексами рассматриваемого типа могут образоваться и ионные ассоциаты типа комплексных кислот. Например, неорганическая часть смешанного соединения, AuCl3 , - это координационно ненасыщенное соединение. Возможны два пути его превращения в координационно насыщенный комплекс:
Выбор пути можно предсказать пользуясь принципом ЖМКО. Золото (III) - мягкий ион, комплексообразование должно преимущественно протекать с более мягким лигандом. Используем обобщенный ряд жесткости лигандов: F > O > N > Cl > Br > I > S. Если L - мягкий серосодержащий экстрагент, более мягкий, чем хлорид-ион, комплексообразование должно пойти по пути I с образованием смешанного комплекса. Если L - жесткий кислородсодержащий экстрагент, хлорид в качестве лиганда будет иметь перед ним предпочтение и комплексообразование должно было бы пойти по пути II с образованием ионного ассоциата. В соответствии с этим серосодержащими нейтральными экстрагентами золото из хлоридных растворов экстрагируется всегда только в виде AuCl3L, а кислородсодержащими экстрагентами, например кетонами, - всегда или почти всегда в виде HAuCl4 . Другая ситуация возникает в случае жестких металлов. Из хлоридных растворов самые жесткие металлы должны хорошо экстрагироваться кислородсодержащими экстрагентами в виде смешанных комплексов, поскольку кислород стоит в ряду жесткости левее хлора.
Следует заметить, что серосодержащими экстрагентами жесткие металлы вообще в большинстве случаев не экстрагируются. Во-первых, это следует из принципа ЖМКО (неблагоприятное сочетание жесткой кислоты - иона металла и мягкого основания, в качестве которого выступает экстрагент). Во-вторых, из-за низкой способности этих экстрагентов к протонизации. В результате не могут образоваться ни смешанные комплексы, ни ионные ассоциаты.
ДЛЯ ЧЕГО НУЖНА ЭКСТРАКЦИЯ
Экстракция, в частности жидкостная, используется для абсолютного и относительного концентрирования, а также для разделения смесей элементов. Абсолютное экстракционное концентрирование заключается в увеличении концентрации вещества за счет его перевода из большого объема водной фазы в меньший объем органической. Оно возможно, если коэффициенты распределения элементов достаточно велики. Из 500 мл воды при большом коэффициенте распределения можно собрать микроэлементы в 5-10 мл органической фазы. Это быстрее и удобнее, чем упаривать воду до такого же объема. Относительное концентрирование увеличивает отношение между микроэлементами и главными компонентами. Относительное концентрирование - это разделение компонентов при резко различающихся их концентрациях. Основная цель относительного концентрирования - замена матрицы, мешающей определению, на подходящий коллектор, чаще всего меньшей массы. В зависимости от цели осуществляют индивидуальное и групповое концентрирование элементов. Концентрирование проводят либо экстрагируя матрицу, либо выделяя экстракцией микроэлементы. Если матрица включает несколько элементов, образующих сложные соединения (геологические и биологические объекты), то лучше экстрагировать микроэлементы. Иногда нет необходимости отделять матрицу полностью. В этом случае уместен термин "обогащение".
Разделение смесей элементов осуществляют прежде всего с помощью избирательных экстрагентов. Например, не составляет труда отделить ртуть и висмут в виде дитизонатов от циркония и алюминия, поскольку ни цирконий, ни алюминий с дитизоном вообще не реагируют. Более типичен случай, когда разделяемые элементы в принципе экстрагируются все, но неодинаково. В этом случае используют другой прием разделения, в основе которого лежит варьирование концентрационных условий: pH, концентрации компонентов системы, включая экстрагент. Разделение достигается также изменением состояния окисления элементов. Например, при разделении галлия и железа аминами эффект достигается восстановлением железа до неэкстрагируемого двухвалентного состояния. Галлий при этом переходит в органическую фазу. Для улучшения разделения при экстракции в водную фазу вводят маскирующие агенты.
Селективность экстракции в некоторых случаях повышают, используя в качестве экстрагента комплекс экстракционного реагента с каким-то другим металлом. Классический пример - экстракция меди диэтилдитиокарбаматом свинца в органическом растворителе. Вытеснить металл, находящийся в исходном комплексе, может только тот элемент, константа экстракции которого больше константы экстракции вытесняемого элемента (для ионов металлов с одинаковыми зарядами). Применяют также и групповое экстракционное концентрирование микроэлементов. Обычными реагентами для такого концентрирования являются 8оксихинолин, дитиокарбаматы, дитизон.
ЗАКЛЮЧЕНИЕ
Проблема разделения смесей и выделения в чистом виде индивидуальных химических соединений имеет огромное практическое значение. В последние десятилетия интерес к этой проблеме усилился в связи с развитием металлургии цветных и редких металлов, полупроводниковой техники. Усовершенствование методов разделения и концентрирования стимулируется также развитием других областей производства, таких, как нефтяная, химическая, фармацевтическая промышленность.
Основными преимуществами экстракционного метода являются высокая избирательность и чистота разделения, возможность работы как с большими, так и с самыми малыми концентрациями, отсутствие загрязнений продуктов, легкость технологического и аппаратурного оформления, возможность осуществления непрерывного процесса, автоматизации и, наконец, высокая производительность. Эти особенности делают экстракционный метод перспективным для применения в различных отраслях промышленности.
Области применения экстракции быстро расширяются. В настоящее время можно назвать аналитическую химию, радиохимию, ядерную технологию, технологию цветных и редких металлов. Кроме того, необходимо отметить большое значение экстракции для препаративных и аналитических целей в научных исследованиях, например при изучении процессов комплексообразования и состояния веществ в растворах. Развитие экстракционных методов достигло такой ступени, что в настоящее время можно экстрагировать любой элемент или разделить любую пару элементов путем применения тех или иных экстракционных систем или выбора соответствующих условий экстракции. Для прогнозирования экстракционной способности различных соединений используются достижения термодинамики, координационной химии, теории растворов, органической химии. Поэтому изучение экстракционных систем способствует развитию химии в целом.
ЛИТЕРАТУРА
1. Золотов Ю.А., Кузьмин Н.М. Концентрирование микроэлементов. М.: Химия, 1982. 288 с.
2. Золотов Ю.А. Экстракция в неорганическом анализе. М.: Изд-во МГУ, 1988. 82 с.
3. Золотов Ю.А. Экстракция внутрикомплексных соединений. М.: Наука, 1968. 313 с.
4. Харитонов Ю.А. Комплексные соединения // Соросовский Образовательный Журнал. 1996. ╧ 1. С. 48-56.
5. Будников Г.К., Троепольская Т.В., Улахович Н.А. Электрохимия хелатов металлов в неводных средах. М.: Наука, 1980. 192 с.
6. Москва В.В. Понятие кислоты и основания в органической химии // Соросовский Образовательный Журнал. 1996. ╧ 12. С. 33-40.
5. Экстракция в органической химии
Экстракция твердых и жидких веществ
Экстракция - процесс разделения смеси твердых или жидких веществ с помощью избирательных растворителей (экстрагентов). Физическая сущность экстракции состоит в переходе извлекаемого вещества из жидкой или твердой фазы в фазу жидкого экстрагента при их взаимном соприкосновении.
Экстракция органическими растворителями - один из наиболее эффективных и универсальных методов разделения, концентрирования и очистки веществ.
В простейшем случае для экстракции растворенного вещества из воды к водному раствору прибавляют органический растворитель (экстрагент), практически не смешивающийся с водой, и смесь взбалтывают для ускорения распределения растворенных веществ между двумя жидкостями. По установлении межфазного равновесия, что обычно достигается после взбалтывания в течение 2-5 мин, взбалтывание прекращают, и жидкие фазы вновь расслаиваются.
При экстракции в делительной воронке нижний слой жидкости можно аккуратно слить и таким образом разделить два растворенных вещества. Если плотность экстрагента больше плотности воды, то он образует нижний слой, а если меньше - верхний.
Предположим, что исходный водный раствор содержал два растворенных вещества А и Б. Если сродство органического растворителя к одному из них намного больше, чем сродство к нему воды, то это вещество полностью или почти полностью перейдет из водной фазы в органическую. Распределение растворенного вещества между двумя жидкими фазами определяется законом распределения: согласно ему отношение концентрации вещества, которое растворено в двух несмешивающихся и находящихся в равновесии жидких фазах, при определенной температуре - величина постоянная, называемая концентрационным коэффициентом распределения Dc:

Если разность Dc экстрагируемых веществ в двух несмешивающихся растворителях достаточно велика, то их можно разделить простой однократной экстракцией; в ином случае применяют многократную простую или же дробную экстракцию.
В отличие от большинства других методов очистки веществ экстрагирование органическими растворителями из водных растворов чаще всего осуществляют при относительно невысокой температуре, а иногда при охлаждении, что особенно удобно при работе с термически нестойкими веществами. Эффективность экстракции в системе твердое вещество - жидкость определяется прежде всего растворимостью и скоростью перехода твердого вещества в растворитель.
Растворимость твердого экстрагируемого вещества можно регулировать, подбирая растворитель с таким расчетом, чтобы в раствор переходило преимущественно очищаемое вещество, а загрязнения оставались в твердой фазе. Скорость перехода вещества из твердой фазы в раствор определяется, в основном, скоростью проникновения экстрагента в твердую фазу, диффузии экстрагируемого вещества в жидкости и удаления вещества с поверхности твердое тело - жидкость (влияние температуры и перемешивания).
В отличие от системы жидкость - жидкость, на границе твердой и жидкой фаз равновесия практически не наступает. Основная задача, возникающая при экстракции твердых веществ (смол, жиров, растительных продуктов), заключается в максимальном ускорении приближения системы к состоянию равновесия, что достигается увеличением поверхности твердой фазы (измельчение), постоянной подачей свежего растворителя на границу фаз, перемешиванием или противотоком.
В настоящем разделе рассматриваются лишь простейшие методы экстракции, обычно используемые в практике работы препаративных и аналитических лабораторий. Более подробные сведения о методах экстракции (дробной, противоточной) можно найти в рекомендуемой литературе.
Экстракция твердых веществ
Выделение и очистка твердого вещества может осуществляться однократной или многократной экстракцией.
При однократной экстракции твердое тонкоизмельченное вещество при перемешивании нагревают с растворителем, (экстрагентом) в колбе с обратным холодильником, после чего экстракт отфильтровывают, испаряют растворитель и выделившееся твердое вещество очищают перекристаллизацией.
При многократной экстракции экстрагируемый материал помещают непосредственно в экстракционный сосуд (насадку), снабженный промежуточным дном из пористого стекла, либо в специальный патрон из фильтровальной бумаги. Пары растворителя из перегонной колбы поступают в обратный холодильник, и сконденсированный растворитель попадает на экстрагируемое вещество в патроне. Когда экстракционный сосуд наполнится до сгиба сливной трубки, экстракт сбрасывается по сифону в перегонную колбу, и процесс экстрагирования повторяется. В подобных экстракторах непрерывного действия экстракт на поверхности экстрагируемого вещества все время обновляется.
При экстракции твердых веществ, смол, масел и растительных продуктов органическими растворителями часто применяют аппарат Сокслета (рис. 171). Эффект экстракции в аппаратах Сокслета зависит, главным образом, от конструкции насадки. Выпускаются лабораторные стеклянные насадки для экстракции твердых веществ с конусными шлифами типа НЭТ, со впаянным стеклянным фильтром (типа НЭТФ) и со вставным стеклянным вкладышем (типа НЭТВ), с конусными шлифами (рис. 172).
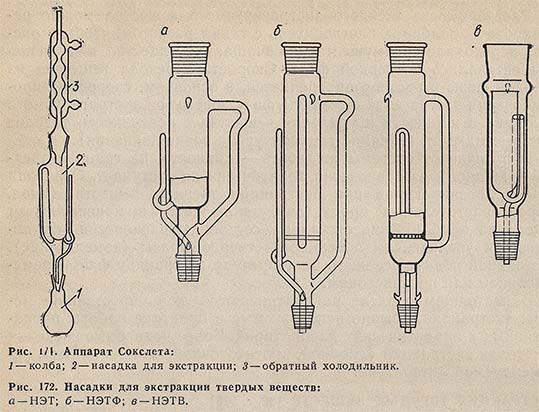
На лабораторном штативе укрепляют колбу с соответствующим номером КШ муфты, присоединяют насадку и обратный холодильник с КШ 45/40 и проверяют герметичность соединений установки.
Размер насадки выбирают так, чтобы патрон с экстрагируемым веществом заполнял приблизительно 2/3 ее объема. Навеску экстрагируемого вещества помещают в стеклянный вставной вкладыш насадки НЭТВ (при малой массе навески) или в патрон из фильтровальной бумаги - при использовании насадок НЭТ и НЭТФ. Патроны с твердым веществом вводят в насадки и наливают растворитель до тех пор, пока он не начнет стекать по сифонной трубке в колбу. Когда растворитель стечет полностью, его добавляют еще один раз, а затем присоединяют обратный холодильник, пускают в него холодную воду, погружают колбу в водяную баню и начинают нагревать. Продолжительность нагревания устанавливается опытным путем.
Если экстрагируемое вещество окрашено, то окончание экстрагирования определяется моментом, когда жидкость в насадке станет бесцветной. Если экстрагируемое вещество бесцветно, можно периодически отбирать пробы раствора из насадки при помощи капиллярной трубки, которую вводят через холодильник. Пробу испаряют на часовом стекле. Если на стекле не образуется матового пятна или пленки, экстракцию можно считать законченной. Разбирая установку, следует прежде всего отключить холодильник от водопроводной сети, затем осторожно снять холодильник и дать стечь экстрагенту из насадки в колбу, для чего насадку наклоняют так, чтобы жидкость переливалась через отводную трубку.
Полученный экстракт выпаривают до минимального объема и завершают испарение в вакуум-эксикаторе.
Учитывая, что аппарат Сокслета часто работает много часов подряд, а растворитель - горючая жидкость, необходимо обеспечить безопасный обогрев; наиболее надежна воздушная баня с ламповым нагревом.
Экстрагирование твердых веществ водой или водными растворами принято называть выщелачиванием. Обычно выщелачивание или водную экстракцию проводят в стеклянном или фарфоровом стакане при энергичном перемешивании. Затем, прекратив перемешивание, дают жидкости отстояться, сливают прозрачную жидкость на бумажный фильтр, а к не растворившейся части вещества снова добавляют растворитель и повторяют операцию до полного извлечения нужного вещества. Окончание экстракции определяют по специфической реакции извлекаемого вещества.
Экстракция веществ из растворов
Простейший вид экстракции - встряхивание раствора вещества в определенном растворителе с другим растворителем, не смешивающимся с первым. Растворитель, служащий экстрагентом, должен лучше растворять экстрагируемое вещество, чем растворитель, из которого это вещество извлекается.
Для экстракции органических веществ из водных растворов пользуются делительной воронкой, в которой смешивают водный раствор вещества (около 1/2 объема воронки) с экстрагентом (1/5 - 1/3 объема воронки). Воронку закрывают пробкой и энергично взбалтывают в течение нескольких секунд, придерживая верхнюю пробку одной рукой, а кран на спускной трубке - другой. Затем делительную воронку переворачивают краном вверх и, осторожно открывая кран, выпускают образующиеся пары.
Если экстрагентом служит диэтиловый эфир, давление паров которого при комнатной температуре довольно велико, в делительной воронке создается значительное избыточное давление. После выравнивания давления кран делительной воронки закрывают, встряхивание и периодическое выпускание паров продолжают до тех пор, пока газовое пространство над жидкостью в делительной воронке не будет заполнено парами растворителя и давление не перестанет меняться. Тогда приступают к более продолжительному и энергичному встряхиванию. По окончании встряхивания делительную воронку укрепляют в штативе, приоткрывают пробку и дают жидкости полностью разделиться на два слоя, которые должны быть совершенно прозрачны. Далее открывают кран и спускают водный слой (если плотность экстрагента меньше плотности воды) в приемную емкость. Слой органической жидкости через верхнее отверстие делительной воронки выливают в колбу с притертой пробкой.
Водный раствор снова переносят в делительную воронку, добавляют новую порцию растворителя и повторяют процесс несколько раз.
Для того чтобы определить, закончен ли процесс экстракции, небольшое количество последней порции экстракта выпаривают на часовом стекле и высушивают в вакуум-эксикаторе над Mg(ClO4)2.
Полученные вытяжки собирают в одну колбу и сушат с помощью соответствующего осушающего средства. После сушки вытяжку фильтруют, затем удаляют растворитель (отгонкой, выпариванием в вакууме), а остаток очищают путем кристаллизации, сублимации или перегонки.
Часто, особенно при экстракции водных щелочных растворов, образуются эмульсии, которые с большим трудом разделяются на два слоя. В этих случаях разделение слоев может быть достигнуто одним из следующих приемов: 1) через жидкость в делительной воронке продувают ток воздуха; 2) водный слой насыщают NaCl; 3) добавляют несколько капель поверхностно-активного вещества, понижающего поверхностное натяжение (например, октилового спирта).
Если вещество значительно лучше растворяется в воде, чем в органическом растворителе, экстракция путем взбалтывания в делительной воронке не дает хороших результатов, и тогда приходится применять непрерывную экстракцию.
Химическое разделение веществ
Из раствора смеси веществ в органическом растворителе можно выделить компоненты, используя различие их химических свойств. Это достигается встряхиванием раствора в делительной воронке с водным раствором кислоты, если нужно отделить вещество основного характера (амины, аминокислоты, аминоспирты), либо водным раствором Na2CO3 или NaHCO3, если нужно отделить вещество кислотного характера. Эту операцию выполняют так же, как и обычную экстракцию, хотя она основана не на различной растворимости вещества в разных растворителях, а на его химических свойствах.
Непрерывная экстракция растворов
С помощью экстракторов непрерывного действия - перколяторов - можно извлекать вещество из раствора небольшим количеством растворителя. Растворитель при этом испаряют в колбе, а пары его конденсируют в обратном холодильнике. Конденсат в виде мелких капель, проходя через раствор, постепенно обогащается извлекаемым веществом и стекает через перелив обратно в колбу. Перколяторы, в зависимости от того, легче или тяжелее растворитель экстрагируемого раствора, различаются конструктивно.
На рис. 173, а изображен простейший прибор, применяемый для экстракции жидкости более легким растворителем. Растворитель нагревают в колбе 1. Пары растворителя через трубку 2 попадают в обратный холодильник, и конденсат стекает в воронку 3; через трубку воронки растворитель попадает в нижнюю часть пробирки 4 и, поднимаясь на поверхность раствора, извлекает растворенное в нем вещество. Экстракт через боковую трубку стекает в колбу 1.
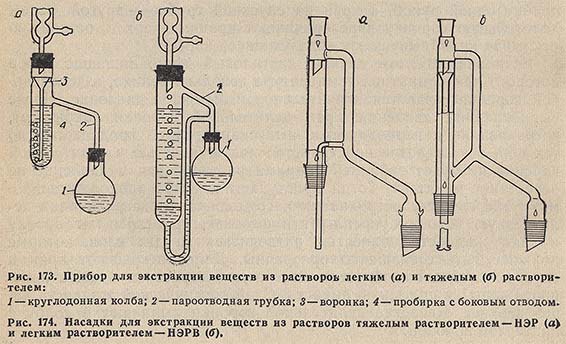
Прибор для экстракции из растворов тяжелым растворителем изображен на рис. 173,6.
Насадки для непрерывного экстрагирования веществ из растворов выпускаются двух типов (рис. 174): НЭР - для экстрагирования тяжелым растворителем; НЭРВ - для экстрагирования легким растворителем.
6. Пробоподготовка в аналитической токикологии
При разработке методик определения лекарственных препаратов в крови и других биологических жидкостях часто возникают проблемы, связанные с мешающим воздействием биологической матрицы на количественное определение аналитов (интерференция). Выявление подобных проблем и поиск путей их устранения весьма актуальны.
Большинство лекарственных веществ имеет высокую степень связывания с
белками крови. Поэтому при изучении токсикокинетики определение лекарств
рекомендуется проводить именно в плазме крови, которая имеет сложную
матрицу, существенно отличающуюся по составу для разных доноров. Согласно
международным рекомендациям Европейского Медицинского Агентства (EMA –
European Medicines Agency) при использовании к примеру масс-спектрометрического
детектирования матричный эффект должен быть оценен на образцах
биологической матрицы не менее шести различных доноров
Большинство современных аналитических приборов, в том числе системы ВЭЖХ-МС, не имеют технической возможности определять малые количества лекарственных
препаратов непосредственно при инжектировании образца. Матрицы
биологических объектов часто очень сложны. Отделить аналит от компонентов
биологической матрицы (плазмы крови) и подготовить его к вводу в прибор –
основные задачи, решаемые с помощью различных подходов пробоподготовки.
Наличие даже самой современной аналитической аппаратуры без
грамотного выбора стратегии процедуры пробоподготовки не может
гарантировать успеха при разработке методики определения лекарственного
препарата в плазме крови. Это связано не только с предельно низкими
концентрациями токсинов, основных активных компонентов лекарственных средств (ЛС) и
их метаболитов, но и с биологической активностью определяемых соединений,
термолабильностью, многокомпонентностью матрицы образца, наличием
оптических изомеров. Именно поэтому правильный выбор процедуры
пробоподготовки часто является ключевым моментом при разработке методики
определения лекарственного препарата в плазме крови.
При проведении практически любого исследования подготовка пробы к
анализу занимает наибольшее количество времени рис. 16. Кроме того, она
является источником более 30% ошибок количественного определения аналитов.
Таким образом, при выборе схемы подготовки проб к анализу необходимо
учитывать не только его надежность и точность, но и такие факторы как простота
эксплуатации, время, необходимое для выполнения процедуры.
Современными тенденциями в пробоподготовке являются миниатюризация,
автоматизация, он-лайн соединение с аналитическим прибором, уменьшение
расхода растворителей и увеличение скорости и безопасности (особенно при
использовании потенциально инфекционных образцов биологического
материала). Минимизация количества стадий при подготовке образцов
необходима для снижения возможных ошибок, что приводит и к повышению
точности. Автоматизированные методы пробоподготовки также весьма
эффективны в плане экономии времени, улучшения воспроизводимости, но при
этом включают и дополнительные расходы.
Кроме того, часто требуется количественное определение не одного
аналита, а целой группы, что имеет место, например, при анализе в плазме крови
действующего лекарственного препарата и его метаболитов. Еще одной
причиной необходимости определения группы аналитов в плазме крови является
оптимизация комплексной терапии, когда пациенту назначают сразу несколько
препаратов, концентрация которых контролируется.
Описаны примеры с одновременным определением 10 аналитов
психотерапевтического действия, 13 противовоспалительных препаратов
40, 15 противовирусных препаратов. Соответственно, метод
пробоподготовки должен учитывать химическую специфику всех аналитов.
Применение метода внутреннего стандарта для количественного
определения лекарственных препаратов в плазме крови уже давно считается
стандартным приемом. Существуют общие стратегии выбора внутреннего
стандарта: он должен иметь близкую по отношению к определяемым веществам
структуру, сопоставимые коэффициенты распределения. Нередко применяют
внутренние стандарты, меченые изотопными атомами. По сути, это –
то же самое анализируемое вещество с массой, отличающейся на один или
несколько Да. Их идентичное химическое поведение позволяет повысить
воспроизводимость и снизить матричный эффект. Однако стоимость подобных
стандартов довольно высока. Авторы предлагают синтезировать меченые
внутренние стандарты непосредственно в лаборатории методом метаболической
инкубации. Другой подход предложен в, а именно: методика выбора
дериватизирующих агентов, аналогичных определяемому аналиту, которые будут
выступать в качестве внутренних стандартов. Метод получил название изотопно-
кодовой дериватизации (ИКД). Применяют реагенты для мечения
функциональных групп в аминах, карбоновых кислотах, тиолах, спиртах и др.
Выделяют три основных подхода при подготовке биологической матрицы к
анализу: осаждение белков, жидкостно-жидкостная экстракция и сорбционное
концентрирование (твердофазная экстракция).
6.1. Осаждение белков
Биологические матрицы представляют собой сложные смеси эндогенных
компонентов, таких как белки, соли, липиды, которые могут взаимодействовать с
аналитами в процессе разделения и хроматографического определения. Белки, в
значительных количествах присутствующие в плазме крови, могут необратимо адсорбироваться на хроматографической колонке, что приведет к ухудшению
эффективности и резкому сокращению срока службы колонки.
Наиболее простой и быстрой процедурой удаления белков биологической
матрицы является их осаждение различными реагентами. Наиболее
распространенными осадителями являются такие органические растворители как
ацетонитрил, метанол, их смесь, добавки в органические
растворители кислот и щелочей. Осаждение белков проводят и сильными
кислотами: муравьиной и хлорной. Применение такого приема позволяет
удалить до 98% белков плазмы крови. Процедура осаждения белков является
быстрой, недорогой и простой в исполнении и может быть применена для
широкого спектра аналитов.
Осаждение белков может также использоваться для образцов плазмы или
сыворотки крови как стадия очистки, предваряющая жидкостно-жидкостную
экстракцию или сорбционное концентрирование. После осаждения
белков проводится центрифугирование и отделение надосадочного слоя, который
и подвергается анализу. Экспрессность является одним из основных преимуществ
данного подхода.
Однако, если требуются низкие пределы обнаружения аналитов,
приходится проводить стадию концентрирования. Для этого растворитель из
надосадочной жидкости выпаривают досуха и содержимое перерастворяют в
меньшем объеме перед вводом в колонку.
Основным недостатком осаждения белков считают недостаточную степень
извлечения. Так, ряд авторов отмечают, что степень извлечения аналита при осаждении
белков обычно не превышает 60%, что вызвано соосаждением аналита с белками
плазмы крови и сложностью биологической матрицы. Кроме того, эффекты
влияния остаточных компонентов после осаждения белков (матричные эффекты)
выявлены в ряде масс-спектрометрических исследований с
электрораспылительной ионизацией. В основном, это подавление
сигнала аналита, приводящее к недостаточной чувствительности и
несоответствию значений повторяемости и правильности экспериментов установленным валидационным требованиям. Именно эти недостатки
ограничивают использование процедуры осаждения белков при разработке
методик определения лекарственных средств в биологических жидкостях.
6.2. Жидкостно-жидкостная экстракция
Жидкостная экстракция (ЖЖЭ) – традиционный тип пробоподготовки,
который отличается селективностью извлечения, несложной реализацией и
невысокой стоимостью.
Чаще всего при разработке биоаналитической методики применяют
наиболее простой – распределительный вариант жидкостно-жидкостной
экстракции. Для этого к биологическому образцу, доведенному до
необходимого значения pH, добавляют органический растворитель. После
перемешивания органический слой отделяют, удаляют растворитель
выпариванием и производят растворение в подходящем для анализа растворе.
Следует отметить, что важную роль при выборе экстрагента играют химические
свойства аналита. Существует простой способ управления полярностью аналитов-
кислот и аналитов-оснований с помощью рН. Ионизованные формы являются
гораздо гидрофильнее их нейтральных аналогов. Вследствие этого следует
ожидать большее сродство к органической фазе (высокое значение KD) в случае,
когда аналит находится в нейтральном состоянии, и, соответственно, – слабое
(низкое значение KD), когда молекула аналита полностью ионизована (Рис.17).
Рис.17. Зависимость коэффициента распределения от значения рН среды
При этом имеется и промежуточный диапазон, в котором сродство к той
или иной фазе варьируется с изменением рН для частично ионизованных форм
аналита. Описываемая кривая аналогична кривой титрования. Точка перегиба
такой кривой находится при значении рН, при котором степень диссоциации
составляет 50% - рКа. Кривые зависимостей «коэффициент распределения - рН»
для кислот и оснований качественно являются зеркальными отображениями друг
друга. В случае кислот следует ожидать увеличения сродства аналита к
органической фазе при низких значения рН, а для оснований – при высоких.
Наиболее распространенными экстрагентами являются
трет.бутилметиловый эфир, этилацетат, их смеси с
дихлорметаном или гексаном]. Для переведения определяемого
вещества в форму, необходимую для лучшего извлечения, используют различные
подходы. Чтобы получить более щелочную среду применяют растворы
гидроксида натрия или аммония. Для подкисления плазмы
до необходимого значения рН используют соляную либо муравьиную
кислоту. Широкое применение для этих целей нашли буферы: ацетатно-
аммонийный, фосфатный. Считается, что именно буферы помогают нивелировать различие в кислотности плазмы крови различных доноров, что
улучшает воспроизводимость при последующем анализе.
При всех преимуществах жидкостно-жидкостной экстракции у нее есть и
ряд недостатков. К основным можно отнести трудоемкость, времязатратность и
довольно большое количество манипуляций с образцом.
Тем не менее, эта процедура гораздо более селективна в сравнении с
осаждением белков и позволяет достигать высоких степеней извлечения.
Жидкость-жидкостная экстракция
Эффективным и распространенным методом в химико-токсикологическом анализе является жидкость-жидкостная экстракция. Этот метод применяется при извлечении ее- ществ из крови, лимфы, слюны, перитонеальной жидкости, мочи и промывных вод желудка. Он также используется на втором этапе при изолировании ядовитых соединений из трупного материала.
Жидкость-жидкостная экстракция — это метод выделения, разделения и концентрирования веществ, основанный на распределении растворенного вещества между двумя жидкими несмешивающимися фазами. В токсикологической химии чаще всего одной жидкой фазой является вода, другой — органический растворитель. Органический растворитель должен обладать высокой растворяющей способностью для анализируемого вещества, иметь низкую температуру кипения. Однако выбор органических растворителей невелик. Наиболее часто для этой цели используют диэтиловый эфир и хлороформ. Выбор органического растворителя определяется свойствами анализируемого соединения.
В условиях равновесия отношение концентраций вещества в обеих фазах представляется константой распределения (К), которая не зависит от общей концентрации вещества.
К = [Со] / [Св]
где Со и Св — равновесные концентрации вещества в обеих фазах (органической, водной) в одной и той же форме.
Однако при экстракции могут происходить различные процессы: ассоциация, диссоциация, сольватация, комплексообразование. Практическое значение при химикотоксикологическом анализе имеет отношение общих (аналитических) концентраций экстрагируемого вещества, которое называют коэффициентом распределения (D).
D = Со / Св
где Со и Св — общие концентрации вещества в органической и водной фазе (независимо от формы существования вещества).
Коэффициент распределения определяют экспериментально в реальных условиях. Его значение используют для разработки условий экстракции ядовитого вещества.
Степень извлечения (процент экстракции) при однократной экстракции рассчитывают по формуле:
R = (100 · D) / (D+Vв/Vо)
где R — степень экстракции, %, D — коэффициент распределения, Vв и Vо — равновесные объемы водной и органической фаз.
Эта формула позволяет рассчитать объем органической фазы для определенного процента извлечения вещества при однократной экстракции.
Пример. Найти объем органического растворителя для извлечения 99% вещества из 100 мл водного раствора при однократной экстракции. Коэффициент распределения равен 20.
Из предыдущей формулы находим:
Vо = (R · Vв) / ( (100-99) · 20)
Подставляя указанные значения R и D, находим:
Vо = (99 · 100) / ( (100-99) · 20) = 495 мл
При многократной экстракции удается добиться полной экстракции значительно меньшим объемом органического растворителя. В этом случае расчет степени экстракции проводится по формуле:
R = 100 · [1- (Vв/ (D·Vo+Vв))n]
где п — кратность экстракции.
Для приведенного выше примера при D=20, объеме водной фазы 100 мл достаточно использовать трехкратное извлечение органическим растворителем объемом 25 мл.
R = 100 · [1- (100/ (20·25+100))3]
Таким образом, данным количеством растворителя можно полнее извлечь растворенное вещество, если проводить экстракцию многократно малыми порциями растворителя.
По приведенной выше формуле можно найти необходимую кратность экстракции при заданных условиях с целью достижения требуемой степени экстракции. Из вышеприведенных формул можно найти:
n = [lg (1- (R/100))] / [lg (Vв/ (D·Vo+Vв)]
Используя данные взятого примера VB=100 мл; V0=25 мл; D=20, найдем число экстракций для извлечения 99% вещества:
n = [lg (1- (99/100))] / [lg (100/ (500+100)] = -2 / -0,778 = 2,57 (округлено 3)
Добавление электролитов в водную вытяжку оказывает высаливающее действие за счет понижения растворимости ядовитых веществ в воде. В результате повышается степень их экстракции органическим растворителем.
6.3. Сорбционное концентрирование (твердофазная экстракция)
Общие сведения о сорбционном концентрировании
Метод сорбционного концентрирования (ТФЭ) предложен более 20 лет
назад и подобен колоночной хроматографии. Он основан на специфических
взаимодействиях выделяемого соединения (или мешающих его определению
компонентов матрицы) с сорбентом.
Типичная схема проведения сорбционного концентрирования приведена на
рис.18. Перед началом процедуры сорбент в картридже сухой, поэтому требуется
его специальная подготовка к работе. Кондиционирование сорбента проводят
растворителем, близким по свойствам растворителю пробы.
Так, если проба является водной, то картридж кондиционируют водой. При
использовании неполярных С18-сорбентов при проведении сорбционного
концентрирования из водных образцов перед кондиционированием водой
проводят так называемую «активацию» сорбента небольшим объемом
органического растворителя: ацетонитрилом или спиртом. После
кондиционирования через картридж пропускают исследуемый образец. При этом
иногда пробе придают определенную кислотность, как в жидкостно-жидкостной
экстракции, для перевода аналита в необходимую форму. Чаще всего это
подкисление фосфорной кислотой или добавка фосфатного буфера с
необходимым значением рН
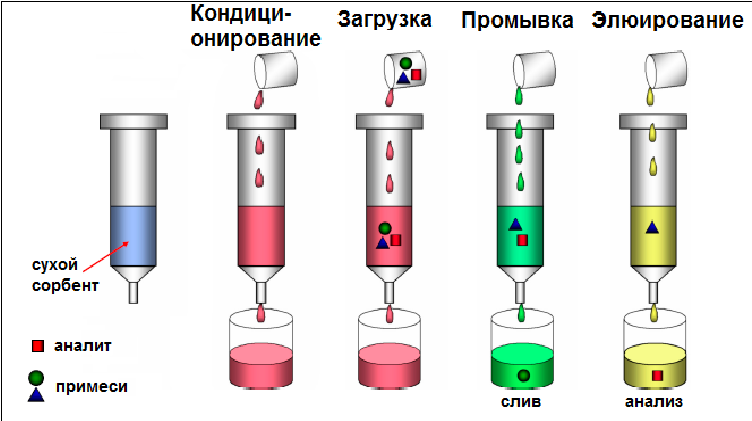
Рис. Общая схема проведения сорбционного концентрирования
Вместе с молекулами аналита на сорбенте удерживаются и многочисленные примеси из биологической матрицы. На стадии промывки применяются растворители (вода, водно-органические смеси, подкисленные/подщелоченные растворы) для элюирования слабо связанных с сорбентом примесей. Завершающей стадией является элюирование аналита с сорбента. Элюирование стараются проводить как можно меньшим объемом растворителя, который, тем не менее, должен быть достаточным для извлечения целевых соединений с картриджа. При этом некоторые примесные компоненты могут оставаться на сорбенте. Чаще всего элюирование проводят метанолом, ацетонитрилом, добавляя к ним гидроксид аммония или муравьиную кислоту в зависимости от аналита. После завершения процедуры извлечения аналита полученный элюат выпаривают досуха и растворяют в подходящем растворителе, при необходимости осуществляя концентрирование. Иногда элюирование проводят подвижной фазой, чтобы сразу подвергнуть элюат анализу в колонке.
Подготовка образцов методом ТФЭ может осуществляться двумя сбособами.
1. Удерживающая ТФЭ
Исследуемое вещество и некоторые мешающие компоненты образца задерживаются на сорбенте при прохождении образца через колонку (картридж для ТФЭ). Примеси, взаимодействующие с сорбентом, удаляются при пропускании через картридж слабого элюента. Затем с помощью сильного элюента, добавляемого малыми порциями, исследуемое вещество также снимается с сорбента. Очищенное, сконцентрированное вещество теперь готово для проведения количественного анализа методом ВЭЖХ или другим аналитическим методом.
2. Неудерживающая ТФЭ
Исследуемое вещество не взаимодействует с сорбентом, находящимся в патроне, в то время как все мешающие компоненты осаждаются на сорбенте. Полученный элюат затем можно сконцентрировать при помощи упаривания или отдувки растворителя и исследовать любыми доступными аналитическими методами.
В ТФЭ используются три основных режима разделения: обращённо-фазовый, нормально-фазовый, ионный обмен.
Обращенно-фазовый режим разделения - экстракция неполярных соединений из водных образцов за счёт Ван дер Ваальсовых взаимодействий.

Нормально-фазовый режим разделения - экстракция полярных соединений из неполярных органических растворителей за счёт водородных связей, диполь - дипольных и π-π взаимодействий.

Ионообменный режим разделения - экстракция заряженных соединений из органических или водных растворов с низкой ионной силой за счёт кулоновских взаимодействий.

| Метод ТФЭ | Обращенно-фазовый | Нормально-фазовый | Сильный ионный обмен | |||
| Сорбент | SDB, C18, C8, PH, CN | Silica, Florisil, NH2, CN | SAX (анионный обмен), SCX (катионный обмен) | |||
| Свойства исследуемого вещества | Слабо полярные (или неполярный), гидрофобные, электрически нейтральные соединения. | Фармацев- тические препараты. Пестициды, гербициды. | Умеренно или сильно полярные, электрически нейтральные соединения. | Пестициды. | Электрически заряженные вещества. | Анионный обмен: анализ веществ, обладающих кислотными свойствами. Катионный обмен: анализ лекарственных препаратов. |
| Образец/ матрица | Водорастворимые вещества в буферном растворе. | Биологические жидкости. Вода. | Неполярные или умеренно полярные органические растворители. | Гексан, хлороформ, сложные эфиры, толуол или метиленхлорид. | Водные буферные растворы низкой ионной силы (с концентрациями не более 30 ммоль) и точно подобранным значением рН | Биологические жидкости плюс буферный раствор. |
| Стадия кондицио- нирования | 1. Сольватация полярными органическими растворителями 2. Уравновеши- вание добавкой воды или буферного раствора | 1. Метанол 2. Вода или буферный раствор | 1. Сольватация полярными органическими растворителями (проводится не всегда) 2. Уравновеши- вание растворителем равным по элюирующей способности с наносимым раствором образца с учетом матрицы | 1. Метанол (не во всех случаях) 2. Гексан или хлороформ | 1. Кондициони- рование полярным органическим растворителем 2. Уравновеши- вание буферным раствором с низкой ионной силой и точно подобранным рН | 1. Метанол 2. 25 mM Tris-0Ac, pH 7 |
| Стадия промывки | Водный буферный раствор с содержанием от 5 до 50% полярного органического растворителя. | Метанол:вода (1:9) | Неполярные органические растворители с низкой концентрацией (от 1 до 5%) слабо полярных органических растворителей | Гексан с 1% тетрагидро- фарана, этилацетат, ацетон, ацетонитрил или изопропанол. | Водяной буфер с низкой концентрацией солей с органическим растворителем или без органического растворителя. | Анионный обмен: Буферный раствор pH7:метанол (50:50) Катионный обмен: 1. Буферный раствор pH 6 2. 1M уксусной кислоты 3. Метанол |
| Стадия элюирования | Полярные или неполярные органические растворители с водой или без воды, буферный раствор, сильная кислота или основание | Метанол: ацетонитрил: HCl (4:4:2) | Неполярные органические растворители с высокой концентрацией (от 5 до 50%) средне и сильно полярных органических растворителей | Гексан с 10% тетрагидро-фарана, этилацетат, ацетон, ацетонитрил или изопропанол. | Анионный обмен: гексан:этилацетат (75:25) + 1% ледяной уксусной кислоты Катионный обмен: метанол + 5% NH3 | |
Для достижения требуемых результатов свойства сорбента для
сорбционного концентрирования должны отличаться от свойств
хроматографической стационарной фазы. Например, сорбент смешанного типа
или ионообменный для ТФЭ используется в сочетании с хроматографической
колонкой С18. Такая комбинация увеличивает шансы на удаление нежелательных
примесей.
Часто для заполнения картриджей используются сорбенты на основе
силикагеля с иммобилизованными катионными или анионными функциональными группами. Одним из важнейших достоинств таких сорбентов
является высокая скорость установления равновесия процессов сорбции и
десорбции, позволяющих работать при достаточно быстром пропускания
анализируемой пробы. Другим их преимуществом является постоянство объема
сорбента при контакте с органическими и водно-солевыми растворами. Сорбенты
на основе силикагеля не требуют длительного предварительного набухания и,
после проведения кратковременной активации и кондиционирования (либо
регенерации) снова готовы к работе.
HLB-картриджи представляет собой модифицированный полистистирол,
обладающий одновременно свойствами гидрофильности и липофильности.
Аббревиатура HLB (Hidrophylic-Lipophylic Balance) в названии сорбента отражает
два его важных уникальных свойства: способность оставаться увлажненным
водой и способность удерживать широкий спектр полярных и неполярных
молекул. Это один из наиболее популярных видов сорбентов для сорбционного
концентрирования при анализе лекарственных препаратов в плазме крови.
В настоящее время техника сорбционного концентрирования может быть
реализована как в виде отдельных картриджей, так и в автоматизированном
96-луночном формате.
Для ТФЭ характерны более широкие возможности варьирования типов
взаимодействий образца с сорбентом и элюентом, чем для жидкостной
экстракции, вследствие чего появляются возможности более селективного и количественного концентрирования и/или извлечения аналитов с участием
специфических взаимодействий (ионообменные механизмы, сродство к
органической фазе.
Кроме того, следует отметить, что при валидации биоаналитической
методики применение сорбционного концентрирования практически полностью
обеспечивает соответствие необходимым требованиям по повторяемости и
правильности. Тем не менее, у этой процедуры есть и ряд недостатков,
основными из которых являются трудоемкость и относительная дороговизна.
Сверхсшитые полистиролы – уникальный нанопористый сорбционный
материал. В основе получения сверхсшитых полистиролов лежит интенсивное
связывание цепей полистирола жесткими мостиками. Три фундаментальных
принципа образуют концепцию формирования сверхсшитых полимерных сетей. Прежде всего, связывание цепей полистирола должно быть статистически
гомогенным, то есть сшивающие мостики должны распределяться случайным
образом в конечной сети. Это достигается за счет однородности исходной
системы, содержащей длинные полимерные цепи и сшивающий агент.
Согласно второму принципу полимер должен оставаться в
сольватированном состоянии на протяжении всей реакции. Это значит, что
используемый растворитель должен быть термодинамически «хорошим» (энергия
взаимодействия растворителя с полимерными цепями превосходит энергию
взаимодействия цепей между собой) как для исходных цепей, так и для конечной
полимерной сетки. Для выполнения этого условия необходимо, чтобы
сшивающий агент не изменял химической природы полимера, то есть имел
близкие к нему состав и полярность. И, наконец, образующаяся структура
полимера должна быть конформационно жесткой. Это достигается путем
48
интенсивного связывания довольно подвижных цепей полистирола при помощи
негибких сшивающих агентов.
Отличительная способность сверхсшитых полистиролов набухать под
действием любых органических жидкостей является следствием основного
принципа формирования сети. Полимер получают за счет фиксации конформаций
сильно сольватированных цепей полистирола при введении большого числа
негибких связей между ними. Таким образом, в образующейся сети практически
отсутствует внутреннее напряжение. При высушивании полимера происходит его
сжатие, что вызывает сильное внутреннее напряжение. Поглощение любой
органической жидкости приводит к расширению сети и возвращению ее в
термодинамически выгодное состояние.
Из-за своей структуры сверхсшитые полистиролы имеют развитую
поверхность, в то же время способны выдерживать механическое воздействие и
взаимодействовать с любой органической жидкостью независимо от ее природы и
термодинамических характеристик. Основное преимущество такого типа
сорбентов заключается в том, что его поры доступны для низкомолекулярных
соединений и при этом недоступны даже для низкомолекулярных белков. По этой
причине не требуется никакой дополнительной пробоподготовки: плазма,
сыворотка крови или моча могут быть нанесены на картридж непосредственно.
Благодаря этому сверхсшитые полистиролы обеспечивают высокую
эффективность при анализе различных объектов, включая биологические.
6.4. Последние достижения в области пробоподготовки плазмы крови к анализу при определении ксенобиотиков
При анализе литературных источников, посвященных различным методам пробоподготовки плазмы крови, выделяя их преимущества и недостатки, можно составить обобщенную схему вариантов пробоподготовки (Рис.).
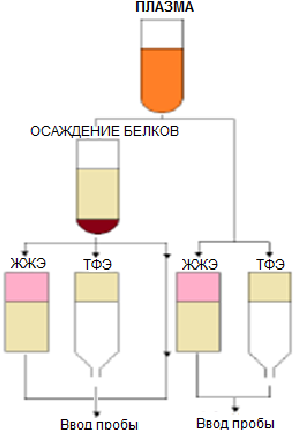
Рис. Обобщенная схема вариантов пробоподготовки плазмы крови при анализе лекарственных препаратов. Выбор стратегии пробоподготовки зависит как от химических свойств аналита, так и от умения исследователя оптимизировать процесс. Одними из современных тенденций в пробоподготовке являются миниатюризация, автоматизация, он-лайн пробоподготовка. Миниатюризация реализована в установках для микротвердофазной экстракции (МТФЭ). Она обычно выполняется с помощью волокон и капиллярных трубок с покрытием соответствующей неподвижной фазой. Экстракции или адсорбция аналитов происходит на внешней поверхности волокна. В другом варианте картридж для МТФЭ представляет собой сменную иглу, которая изнутри заполнена определенным адсорбентом. Адсорбент находится в микрокартридже в верхней части иглы (Рис.).
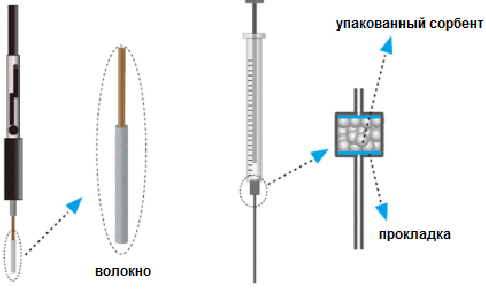
Рис. Примеры реализации МТФЭ. Важным преимуществом МТФЭ является минимальная времязатратность. В распространенных методах экстракции – жидкостно-жидкостной экстракции и при сорбционном концентрировании – процедура извлечения аналитов продолжается, по крайней мере, 15-20 мин. В отличие от них, в методе МТФЭ экстракция занимает на порядок меньшее время, ~ 1-2 мин. Для анализа биологических образцов, где объем пробы часто ограничен, МТФЭ подходит идеально: большое количество образца не требуется. Автоматизация пробоподготовки реализуется как для осаждения белков, так и для экстракций обоих видов. Автоматизация позволяет избежать грубых ошибок, увеличить производительность и надежность анализа, уменьшает влияние аналитика на результаты. Одним из решений является он-лайн пробоподготовка. Она реализована как для ЖЖЭ с использованием мембран, так и для ТФЭ. Методы он-лайн жидкостно-жидкостной экстракции основаны на диффузии аналитов из жидкой матрицы (фаза донора) к жидкой фазе акцептора. Мембрана обычно используется в качестве фазового разделителя или физического барьера. Подвижная фаза проходит через образец, аналиты поступают сразу в колонку. В качестве мембран выступают полые волокна, обеспечивающие низкое сопротивление потоку.
Он-лайн вариант ТФЭ в настоящее время достаточно распространен. Для ввода плазмы крови с определяемым веществом в систему необходимо предварительно провести процедуру осаждения белков. Общая схема проведения он-лайн ТФЭ приведена на рис. Сначала проба попадает в колонку для экстракции, затем подвижная фаза извлекает компоненты пробы и транспортирует их в хроматографическую колонку.
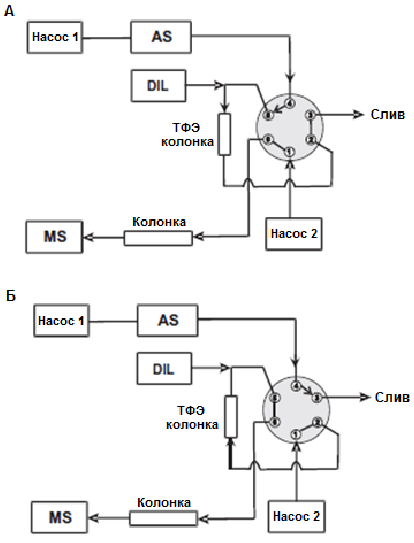
Рис. Общая схема процедуры он-лайн ТФЭ. (AS – автосамплер, DIL – разбавляющий насос, MS – масс-спектрометр). А – процедура ТФЭ, Б – элюирование.
Таким образом, миниатюризация, автоматизация, он-лайн пробоподготовка
позволяют снизить время анализа лекарственного препарата в плазме крови, что
52
особенно актуально, когда в клиническом исследовании задействовано 500-2000
образцов.
Современная процедура пробоподготовки плазмы крови при определении
лекарственных препаратов должна быть не только простой в исполнении,
воспроизводимой и относительно недорогой, но и отвечать международным
требованиям, предъявляемым к биоаналитическим методикам, которые
применяются при проведении клинических исследований.
7. Выделение ДНК из лейкоцитов крови
Выделение общей ДНК из животной клетки не представляет особой сложности, поскольку плазмалемма, ядерная и митохондриальная мембраны «растворяются» в присутствии анионного детергента додецилсульфата натрия (SDS), а сложной клеточной стенки у животных в сравнении, к примеру с растениями, нет. Высвободить ДНК из ДНК-белкового комплекса можно с помощью протеиназ или хаотропных солей. Для этой же цели можно провести как и в случае с бактериями фенольную экстракцию ДНК. Другие вещества при последующем осаждении ДНК спиртом останутся в растворе, и избавиться от них несложно. ДНК можно выделить из любых тканей, клетки которых содержат ядра, но количественный выход из разных тканей может быть различным. Довольно часто для выделения ДНК используют кровь. Лейкоциты крови, в отличие от зрелых эритроцитов, содержат ядра. Общей ДНК, выделенной из 100 мкл цельной крови достаточно для проведения нескольких рестрикций, ПЦР и секвенирования. Митохондриальная ДНК и РНК также присутствуют в получаемом препарате. В приведѐнной ниже методике для освобождения ДНК от белков используется фенол. Под словом «фенол» биологи-молекулярщики часто подразумевают смесь водонасыщенного раствора фенола с хлороформом 1:1, а не кристаллическое вещество. В смеси с хлороформом фенол работает эффективнее, а изоамиловый спирт гасит пенообразование.
1. К 100 мкл цельной крови добавить 2 объема дистиллированной воды до конечного объѐма 0.3 мл. Тщательно перемешать и оставить на 15 минут.
2. Пробу центрифугировать в течение 10 минут, 5000 об/мин. Супернатант (надосадок, надосадочную жидкость) слить.
3. Осадок клеток отмыть двойным объѐмом буфера SSC, добавив в пробирку 200 мкл 1-кратного SSC. Перемешивание аккуратное.
4. Центрифугировать, как в п.2. Супернатант слить.
5. К осадку добавить 54 мкл 0.2М ацетата натрия и 6 мкл 10%-ного раствора SDS. Осадок клеток тщательно ресуспензировать, т.е. перевести в суспензию вновь с помощью микропипетки или вортекса. Инкубировать при 37ºС в течение 0.5–1 ч для прохождения лизиса (разрушения) клеток.
6. Добавить 2 объѐма буфера ТЕ и провести фенольную депротеинизацию образца. Для этого внести в пробирку равный объѐм фенол–хлороформной смеси, хорошо встряхнуть и центрифугировать. Отобрать водную фазу (верхний 11 слой) в чистую пробирку, не захватывая интерфазу – белый осадок на линии раздела фаз.
7. Повторить ту же процедуру, что в п. 6, но со смесью хлороформ- изоамиловый спирт для удаления остатков фенола.
8. К очищенному от белков лизату добавить 1/10 объѐма раствора III, перемешать и высадить ДНК добавив два с половиной объѐма холодного 96% этанола и поместив образец на 1–2 часа в морозильную камеру при –20С. Можно оставить ДНК под спиртом на ночь.
9. Пробу центрифугировать в течение 10 мин при максимальной скорости микроцентрифуги. Супернатант слить. Осадок промыть 70% спиртом.
10. Высушить осадок ДНК на воздухе и растворить в 20 мкл буфера ТЕ.
8. Выделение ДНК из крови с помощью смолы Chelex
Основная процедура выделения включает в себя очистку от металлосодержащих соединений и протеинов с последующим кипячением образца в присутствии Chelex 100, затем супернатант непосредственно добавляют в ПЦР смесь.
1. В центрифужную пробирку на 2 мл вносят 1 мл стерильной дистиллированной воды и добавляют 5 мкл крови или 3 мм2 материала с засохшей кровью. Тщательно перемешивают.
2. Инкубируют при тщательном перемешивании 20 мин при комнатной температуре (лучше всего использовать ротатор для центрифужных пробирок).
3. Центрифугируют при 10 000 g 3 мин и удаляют супернатант, оставляя (чтобы не взмутить осадок клеток или ткань) 20-30 мкл.
4. Добавляют 5% раствор Chelex-100 до конечного объема 200 мкл (Chelex в натриевой форме, 5% раствор в стерильной дистиллированной воде). Перед добавлением смолу перемешивают до гомогенного состояния пипеткой с широким отверстием.
5. Инкубируют при 56 ºС 30 мин и встряхивают в течение 10 сек.
6. Кипятят образец 8 мин при 95 ºС, встряхивают в течение 10 сек. и центрифугируют при 10 000 g 3 мин.
7. Для ПЦР берут из полученного супернатанта 20 мкл
9. Экстракция липидов
В идеальном случае процедура экстракции липидов должна приводить к количественному извлечению клеточных липидов в неизмененном виде. Липидный экстракт не должен быть загрязнен нелипидными веществами, такими как сахара и аминокислоты. Эффективность экстракции липидов в значительной степени зависит от химической природы липидных компонентов и от вида комплексов, которые образуют липиды в клетке с другими классами природных соединений. Известны три основные типа взаимодействия липидов с другими веществами.
Во-первых, ван-дер-ваальсово гидрофобное взаимодействие «нейтральных» или неполярных липидов, таких как эфиры стеринов, глицериды, углеводороды, каротиноиды, которое связывает относительно слабыми нековалентными связями их углеводородные цепи с другими липидами или с гидрофобными участками белков. Такое взаимодействие осуществляется, в частности, в жировой ткани, хиломикронах, комплексах альбумина с жирными кислотами, жировых образованиях микробных клеток и т.п.
Во-вторых, образование водородных связей – электростатическое или гидрофобное взаимодействие, при котором полярные липиды (фосфатиды, гликолипиды, холестерин) образуют связи с белками, как это имеет место в плазматических мембранах, митохондриях, эндоплазматическом ретикулуме и липопротеинах сыворотки.
В-третьих, образование комплексов, в которых жирные кислоты (нормальные или разветвленные) и оксикислоты связаны ковалентными связями (сложноэфирными, амидными или гликозидными) с полисахаридами, как это имеет место в липополисахаридах клеточных стенок бактерий.
Из комплексов, образованных в результате ван-дер-ваальсового гидрофобного взаимодействия, липиды можно экстрагировать относительно неполярными растворителями, такими как этиловый эфир, хлороформ или бензол. Липиды, связанные в мембранах, экстрагируются полярными растворителями, такими как этанол или метанол, разрушающими водородные связи и нарушающими электростатическое взаиможействие белков с липидами. Ковалентно связанные липиды не экстрагируются никакими растворителями; их можно выделить лишь расщепив комплекс с помощью кислотного или щелочного гидролиза.
При выборе метода экстракции необходимо принимать во внимание и химическую природу липидов. Обычно, чтобы предотвратить окисление двойных связей, непосредственно перед экстракцией растворители перегоняют и удаляют из них перекиси. Растворители, применяемые для извлечения высоконенасыщенных липидов, следует дезаэрировать, пропуская через них азот, и затем все операции проводить в атмосфере азота. Липидные экстракты не следует ни упаривать досуха, ни оставлять в упаренном виде на долгое время; извлеченные липиды надо как можно скорее растворять в подходящем растворителе. Температура, при которой проводится экстракция должна быть не выше комнатной (или ниже, если это необходимо) для того, чтобы затормозить окисление и гидролитическое расщепление липидов. Старые способы экстракции кипящим растворителем в аппарате Сокслета должны быть исключены.
Другой фактор, который следует принимать во внимание, особенно при работе с растительными материалами, это ферментативная деградация липидов во время экстракции. Вообще, содержащие спирт смеси растворителей вызывают дезактивацию большинства фосфатидаз и липаз. Более стабильные ферменты разрушаются при 1-2 минутном контакте с кипящей водой или горячим спиртом. Однако, некоторые части растений, как, например, зеленые листья, содержат очень стабильный фермент – фосфолипазу D, которая дезактивируется только при погружении листьев на 1 минуту в кипящую воду или при экстракции кипящим изопропиловым спиртом в течение нескольких минут.
Из вышесказанного следует, что спирт является необходимым компонентом всех смесей, употребляемых для экстракции липидов, так как он разрушает комплексы липидов с белками, растворяет липиды и дезактивирует ферменты, вызывающие расщепление липидов. Однако смест растворителей, содержащие спирт, наряду с липидами экстрагируют содержащиеся в клетках нелипидные вещества, такие как сахара, аминокислоты, соли и т.д. Поэтому неочищенный липидный экстракт нужно обработать так, чтобы удалить все водорастворимые примеси.Наиболее распространенной процедурой такого рода является промывка экстракта водой, приводящая в некоторых случаях к образованию очень стабильных эмульсий. Освобождают липиды от нелипидных примесей, также пропуская неочищенные экстракты через колонки, заполненные целлюлозой или сефадексом, или проводя диализ через специальные мембраны.
Одним из наиболее часто применяемых и эффективных способов экстракции липидов является экстракция по Блайю и Дайэру – упрощенный вариант «классической» методики Фолча. При экстракции по этому методу используют однофазную систему растворителей хлороформ-метанол-вода (1:2:0,8 по объему), которая быстро и эффективно извлекает липиды. Экстракт разбавляют одним объемом воды и одним объемом хлороформа. В результате образуется двухфазная система, нижний слой которой состоит из хлороформа, а верхний – из смеси метанола и воды (1,0:0,9). Водорастворимыне нелипидные примеси переходят в воднометанольный слой, в то время как в хлороформном слое остаются липиды, практически свободные от загрязнений.
Экстракция липидов по Фолчу
10 г тщательно измельченного продукта (с содержанием воды не более 20 %) экстрагируют 3 раза смесью хлороформ-этанол в соотношении 2:1 (50 см3). Первую экстракцию проводят при встряхивании на аппарате в течение 2 ч, две последующие - по 1 ч.
Раствор отфильтровывают в делительную воронку через бумажный фильтр. Остаток на фильтре промывают смесью хлороформ-этанол (4 раза по 10 см3). В делительную воронку добавляют воду (20% от объема экстракта, интенсивно встряхивают и оставляют на ночь до полного разделения хлороформного и водно-спиртового слоев.
Хлороформный фильтрат отделяют от водно-спиртового, переносят в мерную колбу и доводят до метки