Медиаторы воспаления
| Site: | Электронный информационно- образовательный портал ВолгГМУ |
| Course: | Дисциплина Экспериментальная патобиохимия клетки |
| Book: | Медиаторы воспаления |
| Printed by: | Гость |
| Date: | Saturday, 18 May 2024, 3:19 PM |
Table of contents
1. ВВЕДЕНИЕ
Воспаление - типический патологический процесс, местный ответ сложных организмов на повреждение, в ходе которого осуществляется переход белков плазмы и лейкоцитов крови из микроциркуляторных сосудов в очаг повреждения.
Повреждение может быть вызвано разными факторами. Однако характер воспалительного ответа в известной мере не зависит от качества повреждающего стимула. Воспаление развивается по своим собственным (внутренним) законам. Это объясняется тем, что весь ход воспалительного процесса управляется, в основном, эндогенными химическими веществами, которые появляются в очаге повреждения. Вещества эти называются медиаторами (посредниками) воспаления. Ниже дана краткая характеристика некоторых важнейших и наиболее изученных медиаторов воспаления.
2. Источники и химическая природа МВ
Источники медиаторов воспаления
Источниками медиаторов воспаления могут быть:
- белки крови и межклеточной жидкости;
- клетки, в том числе все клетки крови, клетки соединительной ткани, нервные клетки, другие клетки;
- неклеточные элементы соединительной ткани.
Химическая природа медиаторов воспаления
Роль медиаторов воспаления выполняют:
- моноамины; - липиды;
- пептиды; - нуклеозиды и нуклеотиды;
- белки; - протеогликаны.
3. Преформированные и вновь образующиеся медиаторы
Часть потенциальных медиаторов воспаления образуется специальными клетками постоянно, заблаговременно, в отсутствие всякого повреждения. Эти медиаторы накапливаются в определенных “хранилищах”, например, в гранулах тучных клеток, и высвобождаются тотчас после возникновения повреждения. Их называют “преформированными медиаторами”. (типичный пример - гистамин). Другие медиаторы образуются “ex tempore” в ответ на повреждение. Это так называемые “вновь образующиеся” медиаторы. (Типичный пример - простагландины).
3.1. Гистамин
Гистамин - Моноамин гистамин - продукт декарбоксилирования аминокислоты гистидина.

История Изначально гистамин был «молекулой-сиротой»: ученые, проводившие первые исследования, даже не думали, что он может присутствовать в теле человека. Но обо всём по порядку. Эрнест Форно Однако первым антигистаминным средством, вошедшим в клиническую практику, стал антегран (RP 2339): Впоследствии на рынок была выпущена его модификация – пириламин: Стоит также отметить, что из-за недостатка фактического материала первые исследования антигистаминных препаратов проводились достаточно грубо: единственным свойством, определявшим допуск вещества к клиническим испытаниям, являлась способность купировать бронхоспазм у экспериментальных животных. На побочные эффекты вроде сонливости и частичного холинолитического действия никто не обращал внимания. Тип гистаминовых рецепторов, находящихся в бронхах и подвздошной кишке, получил обозначение Н1. Исследователям стало очевидно, что для характеризации и изучения функций Н2-рецепторов (в предыдущих исследованиях они были обнаружены в матке и правом предсердии) нужны новые селективные лиганды. Нужные соединения были созданы в 1965 году. Джеймс Блек, работавший ранее над разработкой лигандов бета-адренорецепторов, решил пойти по пути модификации исходных гормонов. Следуя логике, которой он руководствовался при создании изопреналина и дихлоризопреналина, Блек решил синтезировать различные модификации гистамина и провести анализ их фармакологической активности. Итогом стали два соединения – 5-метилгистамин и N-альфагуанилгистамин, избирательно возбуждавшие Н2-рецепторы. 5-метилгистамин N-альфагуанилгистамин Окончательным итогом его работы стали Н2-блокаторы буримамид и метиамид. Буримамид интересен тем, что он первым полностью блокировал эффекты гистамина в отношении тонуса сосудов. Метиамид же стал отправной точкой для разработки циметидина – Н2-блокатора, использующегося для понижения кислотности желудочного сока и лечения язвенной болезни. Наконец, в 1990х был выделен четвёртый тип гистаминовых рецепторов. В 1996 году Рейбль обнаружил, что увеличение внутриклеточной концентрации кальция в эозинофилах в ответ на введение гистамина можно успешно блокировать тиоперамидом, но не пириламином или циметидином. Это стало доказательством того, что в данном случае эффект являлся следствием возбуждения Н3-рецепторов. Однако в последующих экспериментах с альфа-метилгистамином (избирательным Н3-агонистом) увеличения концентрации кальция в эозинофилах не происходило. Подобным образом были протестированы и другие известные на тот момент Н3-агонисты, однако они не оказывали на эозинофилы такого же эффекта, как чистый гистамин. Это дало исследователям основания утверждать о выделении нового типа гистаминовых рецепторов – Н4. В дальнейшем выяснилось, что Н4-рецепторы находятся главным образом в клетках иммунной системы, и с нарушением их функционирования связали несколько аутоиммунных заболеваний, однако их селективные лиганды до сих пор не нашли клинического применения. У мышей, лишённых Н4-рецепторов, нарушен процесс хемотаксиса иммунных клеток. Также Н4-рецепторы являются единственными, чья структура остаётся не ясной до конца.
Эрготизм, или же Антониев огонь, был описан ещё средневековыми медиками. Это заболевание с довольно интересной симптоматикой: диарея, галлюцинации, судороги, при тяжёлых формах возможно возникновение гангрены. Однако лишь к середине XIX века у врачей появилось относительно реалистичное представление о главной причине эрготизма – паразитическом грибке Claviceps purpurea, он же спорынья. Внимательный читатель спросит: «А причём здесь, собственно, спорынья, если мы говорим о гистамине?» В 1904 году кембриджский аспирант-физиолог Генри Дейл и его научный руководитель Джон Лэнгли были приглашены на работу в лабораторию Генри Уэллкома в Южном Лондоне. Уэллком поставил исследователям задачу выяснить механизмы действия экстракта спорыньи и найти ему новые терапевтические приложения. К их работе подключился Джордж Баргер, который к тому времени уже имел опыт выделения и очистки отдельных соединений из спорыньи, и вместе с Дейлом они начали систематизацию и описание фармакологических свойств выделенных веществ. В 1910 году они выделили из стандартизированного экстракта спорыньи бета-аминоэтилимидазол, имевший структурное сходство с аминокислотой гистидином, но лишённый карбоксильной группы. Впоследствии он получил название «гистамин». Однако позднее выяснилось, что за три года до выделения гистамина из спорыньи это вещество уже было описано в 1907 году Виндхаусом и Вогтом в качестве синтетического аналога гистидина. Это не могло не порадовать Дейла: синтез гистамина оказался намного легче его выделения. Таким образом, Дейл получил источник больших количеств гистамина для последующих исследований его фармакодинамики и фармакокинетики. Первые исследования по изучению действия гистамина проводились на млекопитающих; в процессе выяснилось, что соединение способно вызывать сокращение гладкой мускулатуры матки, бронхов и кровеносных сосудов, также гистамин вызывал усиление секреторной функции слизистых оболочек и увеличение кислотности желудочного сока. Также Дейл в сотрудничестве с Линдлоу отметил существенное сходство между анафилактической реакцией и эффектами, вызываемыми введением больших количеств гистамина подопытным морским свинкам. В 1927 году Дейл доказал существование эндогенного гистамина, обнаружив его в экстрактах печени и лёгких сенситизированных животных.
Спустя десятилетие медики признали связь между гистамином и аллергическими реакциями, и в 1937 году Бове и Штауб из Института Пастера, используя соединения, синтезированные Эрнестом Форно, показали возможность частичного блокирования действия гистамина. Первым антигистаминным препаратом, показавшим нужную активность, был пипероксан.


Со временем выяснилось, что не все эффекты гистамина блокируются введением антигистаминных препаратов. Это натолкнуло учёных на мысль о существовании нескольких типов рецепторов гистамина.
Доказательства этой гипотезы появились уже в 1940-х годах: первые модели действия гистамина были опубликованы Уэллсом в 1945 и Фолкоу в 1948 годах. В качестве доказательства существования нескольких типов гистаминовых рецепторов приводился опытах на кошках, где димедрол лишь частично блокировал вазодилатацию, вызванную введением гистамина.
В 1960 году Тренделенбург в процессе уточнения pA2* для пириламина обнаружил, что для разных органов (сердце и подвздошная кишка) значение pA2 довольно сильно отличается. Впоследствии было установлено, что пириламин в малых дозах блокирует эффекты гистамина в отношении бронхов и мускулатуры подвзошной кишки, однако не оказывает никакого воздействия на тонус матки и правого предсердия.
*pA2 применяется для относительного измерения фармакологической активности соединений; это отрицательный логарифм молярной концентрации антагониста, при которой для получения стандартного эффекта агониста его концентрацию надо увеличить вдвое.

Однако и это ещё не все. В последующих исследованиях выяснилось, что совместное введение Н1- и Н2-блокаторов купировало не все эффекты гистамина, и особенно ярко это проявилось во время опытов на изолированных срезах мозга. В 1983 году Аррандж проводил исследование с меченным изотопной меткой гистамином и выяснил, что буримамид (раннее классифицированный как Н2-блокатор) и импромидин (Н2-блокатор) имели слишком большую разницу в активности: относительно гистаминовых рецепторов, находившихся в срезах коры головного мозга мышей, буримамид оказался в 300 раз более активным. Это дало основание для выделения нового типа гистаминовых рецепторов – Н3. Позже, в 1987 году, Тжечаковский обнаружил похожую разницу в фармакологических активностях буримамида и импромидина относительно активности нервов ауэрбахового сплетения. Его исследования привели к разработке модельных лигандов Н3-рецепторов – агониста альфа-метилгистамина и антагониста тиоперамида. В настоящее время некоторые лиганды Н3-рецепторов рассматриваются как потенциальные лекарства против болезни Альцгеймера, СДВГ и нарколепсии.
Недавно (2012, «Histamine-gated ion channels in mammals?» Fleck M., Biochemical Pharmacology) появились сообщения об ионных каналах каналах хлора, для которых гистамин является специфическим лигандом, однако на сегодняшний день они недостаточно хорошо описаны.
Главный источник гистамина при воспалении у человека - тучные клетки тканей и базофилы крови. Как тучные клетки, так и базофилы - высокоспециализированные клетки, содержащие большое число гранул - округлых, ограниченных мембраной структур, основу которых составляет протеогликановый матрикс. В тучных клетках соединительной ткани человека роль протеогликанового матрикса выполняет гепарин, в базофилах - хондроитин 4-сульфат. Внутри гранул, в среде с низким pH, содержатся в преформированном виде биологически активные вещества, в том числе и гистамин, который удерживается кислыми группами гранулярного матрикса с помощью ионной связи. На долю гистамина приходится около 10% сухой массы гранул тучных клеток.

Значительная часть тучных клеток сосредоточена в коже, в слизистых оболочках верхних и нижних дыхательных путей, желудочно-кишечного тракта, мочеполовых органов, где они располагаются вокруг мелких кровеносных и лимфатических сосудов и в местах скопления нервных окончаний.
Высвобождение гистамина из тучных клеток и базофилов есть сложный процесс, тесно связанный с изменением их гранулярного аппарата - с дегрануляцией. По результатам морфологических исследований, дегрануляция одних гистамин-содержащих клеток, например тучных клеток кожи человека, обусловлена выбросом интактных гранул вместе со всем их содержимым во внеклеточную среду. Дегрануляция других клеток, например тучных клеток слизистой носа и легких, обусловлена слиянием мембран отдельных гранул друг с другом и с цитоплазматической мембраной. В результате образуются сообщающиеся с внешней средой каналы, через которые солюбилизированное содержимое гранул диффундирует во внеклеточное пространство. Во внеклеточной среде, pH которой значительно выше чем внутри гранул, гистамин становится свободно растворимым и легко диффундирует из места своего высвобождения. Дегрануляция тучных клеток и базофилов может быть вызвана различными стимулами (таблица 1).
Таблица 1.
Факторы, вызывающие дегрануляцию тучных клеток
- Иммуноглобулины класса Е (после фиксации антигена)
- Фрагменты комплемента (С3а, С5а)
- Нейропептиды (вещество Р, нейротензин)
- АТФ
- Физические стимулы (вибрация, нагревание, охлаждение)
- Факторы, высвобождаемые клетками, участвующими в воспалении: нейтрофилами, лимфоцитами, тромбоцитами, эндотелиальными клетками, эозинофилами, макрофагами легких
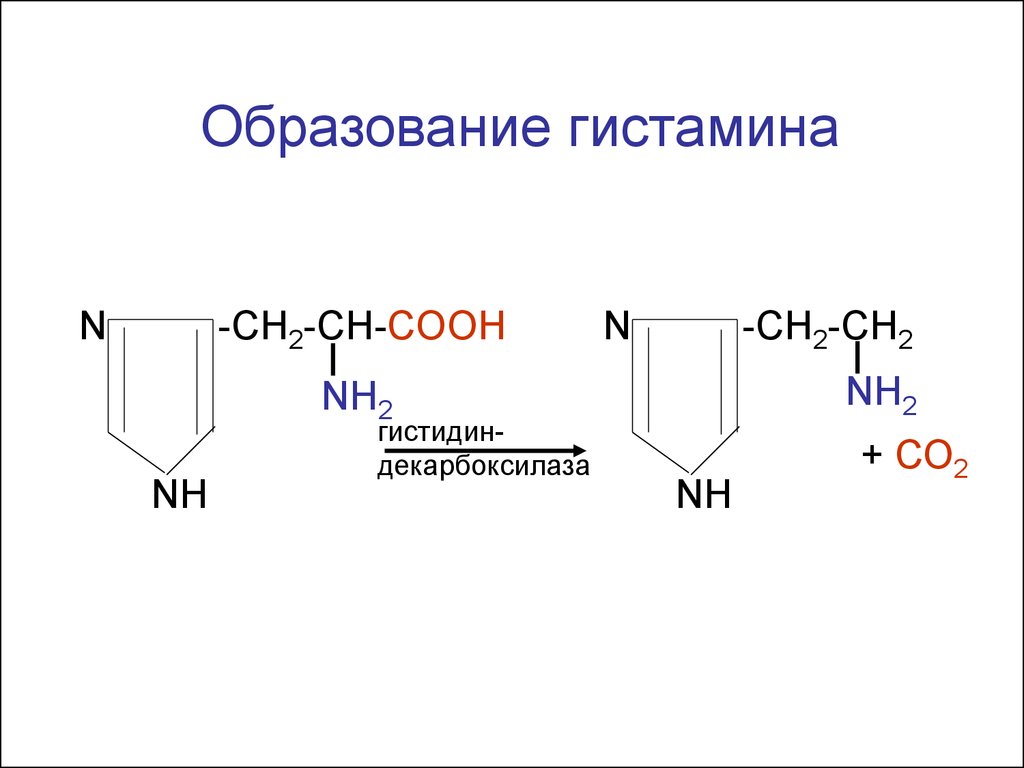
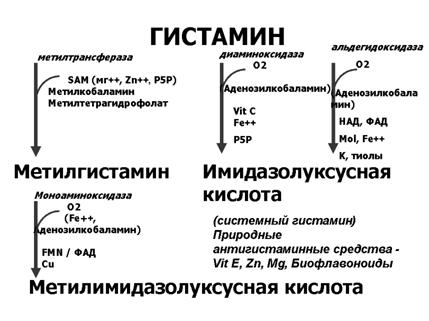
Рисунок 1. Синтез и распад гистамина.
От 50 до 70% свободного гистамина преобразуется ( метаболизируется) N-метилтрансферазой в N-метилгистамин. Фермент N-метилтрансфераза содержится в тонком кишечнике, печени, почках, в других органах, в лейкоцитах. Остальной гистамин метаболизируется диаминоксидазой (гистаминазой) в имидазолуксусную кислоту. Значительные количества гистаминазы содержатся в слизистой кишечника, в плаценте, печени, тимусе, эозинофилах, нейтрофилах.
Рецепторы к гистамину
Действие гистамина на клетки-мишени реализуется через специальные рецепторы. Все изученные и охарактеризованные на сегодняшний день рецепторы гистамина являются метаботропными и осуществляют своё действие через цепь вторичных внутриклеточных мессенджеров.
ПНС: ключевое звено механизма бронхоконстрикции, регулирует процессы вазодилатации, висцеральной ноцицепции, участвует в развитии крапивницы и зуда. ПНС: гладкая мускулатура, эндотелий, чувствительные нейроны.Рецептор Тип Функция Местонахождение Агонисты Антагонисты Н1 Gq – вызывает повышение уровня IP3 и ДАГ. ЦНС: регуляция цикла «сон-бодрствование» (осуществляет переход в состояние бодрствования), регуляция температуры тела, ноцицепции, нейроэндокринного гомеостаза, процессов запоминания и аппетита. ЦНС: туберомаммилярные ядра гипоталамуса, которые имеют проекции к дорсальным ядрам шва и синему пятну. Гистамин, HTFMT, UR-AK49, бетагистин, 2-пиридилэтиламин Дифенгидрамин, лоратадин, цетиризин, фексофенадин, клемастин Н2 Gs – увеличивает уровень цАМФ. Частично участвует в процессе вазодилатации, регулирует секрецию желудочного сока и функции ЖКТ. Париетальные клетки желудка, гладкая мускулатура сосудов. Гистамин, амтамин, бетазол, димаприт, HTFMT, импромидин, UR-AK49 Ранитидин, циметидин, фамотидин, низатидин Н3 Gi – снижает уровень цАМФ. Пресинаптический ауторецептор, ингибирует выброс гистамина, серотонина, норадреналина и ацетилхолина. Регулирует аппетит и частично секрецию желудочного сока. Главным образом находятся в ЦНС. Гистамин, альфа-метилгистамин, ципралисант, иметит, иммепип, имметридин, метимипип, проксифан ABT-239, ципроксифан, клобенпропит, тиоперамид Н4 Gi – снижает уровень цАМФ. Участвует в осуществлении хемотаксиса, развитии зуда и воспаления, выбросе цитокинов. Также играет роль в возникновении болевых ощущений. Базофилы, CD4+ лимфоциты, костный мозг, тимус, тонкая кишка, селезёнка, толстая кишка. Гистамин, 4-метилгистамин, альфа-метилгистамин, OUP-16, VUF-8430 Тиоперамид, JNJ-7777120
Также стоит перечислить меченные изотопными метками специфические лиганды, которые используются для исследования рецепторов: Н1 – [3H]-пириламин, Н2 – [125I]-аминопотентидин, Н3 – [125I]-йодопроксифан, Н4 –[3H]- JNJ7777120
![Схема действия гистамина на уровне тканей и клеток на примере аллергической реакции. [2] Источник: журнал Nature](http://medach.pro/wp-content/uploads/2016/01/fig2_1.png)
Рисунок 2. Функциональные последствия активации гистаминовых рецепторов.
Можно заключить, что основные последствия активации Н1 гистаминовых рецепторов у человека
это:
- сокращение гладких мышц (в том числе мышц бронхиол и желудочно-кишечного тракта);
- увеличение сосудистой проницаемости, прежде всего посткапилярных венул;
- увеличение содержания цГМФ в клетках-мишенях ;
- кожный зуд;
- стимуляция образования простагландинов;
- укорочение времени проведения по атриовентрикулярному
- узлу;
- раздражение чувствительных окончаний блуждающего нерва
- в воздухоносных путях;
- Секреция соляной кислоты в желудке;
- Увеличение секреции слизи эпителием воздухоносных путей;
- Увеличение содержания цАМФ в клетках-мишенях;
- Сокращение мышц пищевода;
- Н1+ Н2 Гипотензия; Покраснения кожи;
- Головная боль; Тахикардия.
Гистамин обнаруживается в очаге воспаления по существу одновременно с возникновением повреждения. Вызывает расширение микроциркуляторных сосудов, увеличивает их проницаемость, стимулирует окончания болевых нервов. Таким образом, гистамин “запускает” острый воспалительный Источник: журнал Nature [2] Когда специфический антиген связывается с FCER1, запускается LYN-зависимое фосфорилирование ITAM-фрагментов и активация протеинкиназ FYN и SYK. Эти активные протеинкиназы, в свою очередь, фосфорилируют LAT (LYN и SYK) и GAB2 (FYN). Фосфорилирование LAT ведёт к активации фосфолипазы С γ (PLCγ), а фосфорилирование GAB2 – к активации фосфоинозитид-3-киназы (PI3K). Их активация ведёт к запуску сигнальных путей, активируемых вторичными мессенджерами – инозитолтрифосфатом (InsP3), диацилглицеролом (DAG) и фосфатидилинозитол-3-фосфатом (PtdIns(3,4,5)P3). Это ведёт к активации протеинкиназы С и выходу Ca2+ из эндоплазматического ретикулума в цитозоль. Выход Ca2+ из ЭПР ведёт к активации белка STIM1, который открывает ионные каналы ORAI1 и TRPC1. Они также повышают концентрацию внутриклеточного Ca2+, впуская кальций из межклеточного пространства внутрь. И, наконец, активированная протеинкиназа С и повышенный уровень Ca2+ являются триггерами для запуска процесса дегрануляции. Созревшие везикулы подтягиваются по микротрубочкам ещё ближе к мембране благодаря коронинам 1А и 1В, происходят множественные слияния везикул одна с другой благодаря связям между t-SNARE и v-SNARE, а после везикулы сливаются с мембраной и их содержимое выбрасывается во внеклеточное пространство. Краткая схема работы трёх типов гистаминовых рецепторов (Н4-рецепторы не показаны, т.к. их механизм действия схож с Н3-рецепторами) Сокращения АА – арахидоновая кислота АС – аденилатциклаза CREB – фактор транскрипции, связывающийся с цАМФ-реактивным элементом DAG – диацилглицерол GC – гуанилатциклаза HVACC – потенциал-зависимый кальциевый ток IAHP – кальций-зависимый калиевый ток Ih — активируемый гиперполяризацией катионный ток Ik, leak – калиевый ток утечки Ins(1,4,5)P3 – инозитол-1,4,5-трифосфат MAPK – митоген-активируемая протеинкиназа NCX – натрий-кальциевый обменник NMDA – N-метил-D-аспартат и его рецептор NO – оксид азота II PKA – протеинкиназа А PLA – фосфолипаза А PLC – фосфолипаза С
ответ.
Как следует из схемы, одним из ключевых моментов в запуске аллергической реакции является высвобождение гистамина вместе с протеазами и цитокинами из тучных клеток (mast cells); также этот процесс называют дегрануляцией. Рассмотрим этот момент поподробнее:![Источник: журнал Nature [2]](http://medach.pro/wp-content/uploads/2016/01/fig3_1-1.png)
FCER1 состоят из нескольких субъединиц: альфа-субъединица ответственна за связь с IgE и антигеном, бета-субъединица содержит активационный тирозинсодержащий мотив иммунорецепторов (ITAM), гамма-субъединица также содержит два ITAМ-фрагмента, соединённых дисульфидной связью.
Ну а дальше вы знаете: покраснение кожных покровов, отёк, повышение секреции слизи, иногда бронхоконстрикция и всё такое.
Стоит также отметить, что в париетальных клетках желудка дела обстоят намного легче: там гистамин просто активирует Н2-рецептор, активация которого ведёт к увеличению количества цАМФ и запуску работы H/K-АТФазы через активацию протеинкиназы А. Н/К-АТФаза (она же протонная помпа) транспортирует ионы водорода против градиента концентрации, что ведёт к синтезу HCl и повышению кислотности желудочного сока.
Участие гистамина в развитии воспаления очевидно при многих болезнях человека, но, в особенности, при аллергических болезнях, язвенной болезни желудка и двенадцатиперстной кишки (см. таблицу 4).
Выше указывали, что появление гистамина в очаге воспаления тесно связано с дегрануляцией тучных клеток. Однако внутри гранул тучных клеток и базофилов содержатся помимо гистамина еще и другие вещества - медиаторы воспаления. Кроме того, сам процесс дегрануляции стимулирует синтез новых медиаторов, источником которых служат липиды мембран активированных тучных клеток и базофилов.
3.2. Протеазы
Внутригранулярные протеазы составляют значительную часть общего белка тучных клеток. Их участие в воспалении обосновывается тем, что применение ингибиторов протеаз заметно снижает повреждение тканей, сопровождающее дегрануляцию тучных клеток. Внутригранулярные протеазы, к числу которых принадлежат триптаза и химаза, осуществляют переваривание базальной мембраны кровеносных сосудов, что приводит к увеличению сосудистой проницаемости, расщепляют протеогликаны соединительной ткани, способствуя эмиграции лейкоцитов, расщепляют продукты распада клеток, активируют факторы роста, способствуют заживлению тканей. В экспериментах “ин витро” показана способность протеаз расщеплять такие протеины плазмы как ангиотензин I и C3 компонент комплемента. В условиях “ин виво” это может приводить к образованию вазоактивных метаболитов
3.3. Протеогликаны
Характерная метахроматическая окраска тучных клеток и базофилов основными анилиновыми красителями обусловлена присутствием в них гранул протеогликанов. Протеогликаны имеют протеиновый сердечник, содержащий множество последовательностей серин-глицин, с которыми связаны ковалентно гликозаминогликановые боковые цепи, состоящие из повторяющихся сульфатированных дисахаридов. В тучных клетках соединительной ткани человека доминирующим протеогликаном является гепарин, в тучных клетках слизистых в базофилах - хондроитинсульфаты. Молекула гепарина содержит несколько сотен сульфатных групп, что
делает ее наиболее негативно заряженной молекулой в организме. В тучных клетках гепарин образует матрикс секреторных гранул, к которому с помощью ионной связи прикреплены многие преформированные, положительно заряженные медиаторы, в том числе гистамин и протеазы. Высвобождаясь из тучных клеток вместе с гистамином и другими медиаторами, гепарин оказывает противосвертывающее действие. Кроме того он способен тормозить активность некоторых лизосомальных ферментов, содействовать прикреплению фибронектина к фибробластам, стимулировать миграцию клеток капиллярного эндотелия.
Замечательная особенность протеогликанов - устойчивость к разрушающему действию протеолитических ферментов. Поскольку протеазы гранул тучных клеток остаются в связи с протеогликанами и после дегрануляции, протеогликаны не только препятствуют интенсивной диффузии протеаз за пределы очага повреждения, но и защищают их от инактивации.
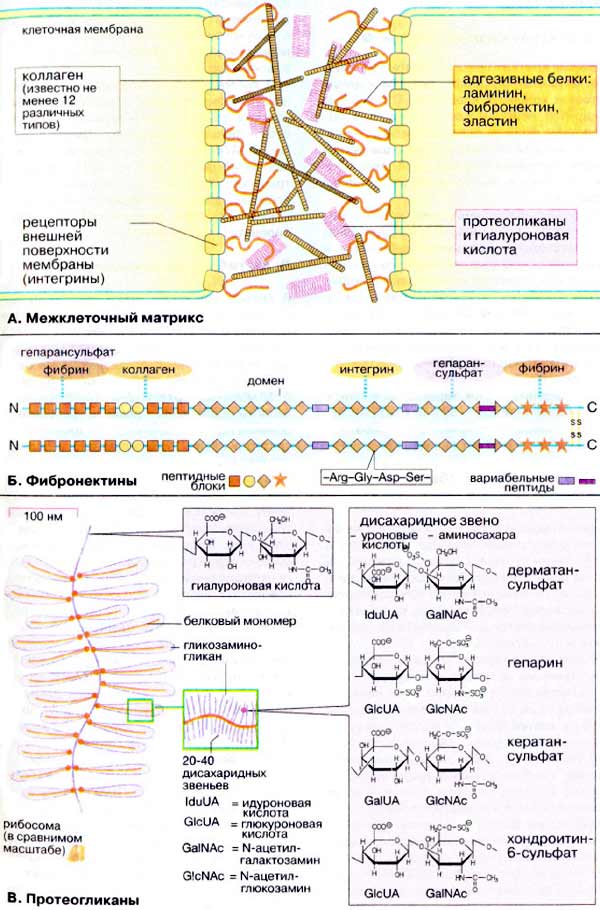
3.4. Хемотаксические факторы
В гранулах тучных клеток в преформированном виде содержатся пептиды, обладающие способностью вызывать положительный хемотаксис эозинофилов. Их называют “факторами хемотаксиса эозинофилов при анафилаксии” (ФХЭ-А). Показано, что свойствами ФХЭ-А обладают кислые тетрапептиды: вал-гли-сер-глу и ала-гли-сер-глу. Помимо хемотаксического действия, которое обнаруживается при достаточно низких концентрациях этих пептидов (10-8М), они увеличивают экспрессию на мембране эозинофилов рецепторов для С4b и C3b компонентов комплемента. К числу префомированных медиаторов гранул тучных клеток принадлежит еще и фактор хемотаксиса для нейтрофилов - белок с молекулярной массой около 600 кД.

3.5. Кинины
Кинины - пептиды, образующиеся в результате ферментативного расщепления специальных глобулинов (кининогенов) плазмы и некоторых других белков. Название “кинины” (“действующие”) указывает на то, что они обладают биологической активностью. Наиболее известный кинин - брадикин (БК) имеет в своем составе 9 аминокислот. Одним из источников БК является высокомолекулярный кининоген плазмы (альфа-2-глобулин).

Высвобождение БК из кининогена происходит благодаря расщеплению двух внутренних связей между аминокислотами в молекуле кининогена под влиянием фермента калликреина. Калликреин присутствует в плазме в неактивной форме и активируется фактором Хагемана. Фактор Хагемана (фактор ХП) - бета-глобулин плазмы - активируется при повреждении вследствие его контакта с отрицательно заряженными поверхностями (контактная активация). Такие поверхности наличествуют у многих органических и неорганических материалов

Брадикинин расширяет микроциркуляторные сосуды, увеличивает сосудистую проницаемость, вызывая образование щелей между эндотелиальными
клетками, вызывает жгучую боль, воздействуя на окончания ноцицептивных нервов. При системном введении снижает периферическое сосудистое сопротивление и понижает кровяное давление. Увеличивает подвижность кишечника, стимулируя сокращение гладкомышечныз клеток. БК и другие кинины
быстро разрушаются под влиянием кининаз (карбоксипептидаз). Существует много оснований полагать, что БК и другие кинины играют
важную роль в воспалительных болезнях суставов, верхних дыхательных путей и легких, при сепсисе, при врожденном ангиноневротическом отеке.
3.6. Комплемент
Система комплемента — комплекс сложных белков, постоянно присутствующих в крови. Это каскадная система протеолитических ферментов, предназначенная для гуморальной защиты организма от действия чужеродных агентов, она участвует в реализации иммунного ответа организма. Является важным компонентом как врождённого, так и приобретённого иммунитета.
В конце XIX столетия было установлено, что сыворотка крови содержит некий «фактор», обладающий бактерицидными свойствами. В 1896 году молодой бельгийский ученый Жюль Борде, работавший в Институте Пастера в Париже, показал, что в сыворотке имеются два разных вещества, совместное действие которых приводит к лизису бактерий: термостабильный фактор и термолабильный (теряющий свои свойства при нагревании сыворотки) фактор. Термостабильный фактор, как оказалось, мог действовать только против некоторых микроорганизмов, в то время как термолабильный фактор имел неспецифическую антибактериальную активность. Термолабильный фактор позднее был назван комплементом. Термин «комплемент» ввёл Пауль Эрлих в конце 1890-х годов. Эрлих был автором гуморальной теории иммунитета и ввёл в иммунологию много терминов, которые впоследствии стали общепринятыми. Согласно его теории, клетки, ответственные за иммунные реакции, имеют на поверхности рецепторы, которые служат для распознавания антигенов. Эти рецепторы мы сейчас называем «антителами» (основой вариабельного рецептора лимфоцитов является прикреплённое к мембране антитело класса IgD, реже IgM. Антитела других классов в отсутствие соответствующего антигена не прикреплены к клеткам). Рецепторы связываются с определённым антигеном, а также с термолабильным антибактериальным компонентом сыворотки крови. Эрлих назвал термолабильный фактор «комплементом» потому, что этот компонент крови «служит дополнением» к клеткам иммунной системы.
Эрлих полагал, что имеется множество комплементов, каждый из которых связывается со своим рецептором, подобно тому, как рецептор связывается со специфическим антигеном. В противоположность этому Борде утверждал, что существует «дополнение» только одного типа. В начале XX века спор был разрешён в пользу Борде; выяснилось, что комплемент может активироваться с участием специфических антител или самостоятельно, неспецифическим способом.
Комплемент — система белков, включающая около 20 взаимодействующих компонентов: С1 (комплекс из трех белков), С2, СЗ, …, С9, фактор В, фактор D и ряд регуляторных белков. Все эти компоненты — растворимые белки с мол. массой от 24 000 до 400 000, циркулирующие в крови и тканевой жидкости. Белки комплемента синтезируются в основном в печени и составляют приблизительно 5 % от всей глобулиновой фракции плазмы крови. Большинство из них неактивны до тех пор, пока не будут приведены в действие или в результате иммунного ответа (с участием антител), или непосредственно внедрившимся микроорганизмом (см. ниже). Один из возможных результатов активации комплемента — последовательное объединение так называемых поздних компонентов (С5, С6, С7, С8 и С9) в большой белковый комплекс, вызывающий лизис клеток (литический, или мембраноатакующий, комплекс). Агрегация поздних компонентов происходит в результате ряда последовательных реакций протеолитической активации с участием ранних компонентов (С1, С2, С3, С4, фактора В и фактора D). Большинство этих ранних компонентов — проферменты, последовательно активируемые путём протеолиза. Когда какой-либо из этих проферментов специфическим образом расщепляется, он становится активным протеолитическим ферментом и расщепляет следующий профермент, и т. д. Поскольку многие из активированных компонентов прочно связываются с мембранами, большинство этих событий происходит на поверхностях клеток. Центральный компонент этого протеолитического каскада — С3. Его активация путём расщепления представляет собой главную реакцию всей цепи активации комплемента. С3 может быть активирован двумя основными путями — классическим и альтернативным. В обоих случаях С3 расщепляется ферментным комплексом, называемым С3-конвертазой. Два разных пути приводят к образованию разных С3-конвертаз, однако обе они образуются в результате спонтанного объединения двух компонентов комплемента, активированных ранее в цепи протеолитического каскада. С3-конвертаза расщепляет С3 на два фрагмента, больший из которых (С3b) связывается с мембраной клетки-мишени рядом с С3-конвертазой; в результате образуется ферментный комплекс ещё больших размеров с измененной специфичностью — С5-конвертаза. Затем С5-конвертаза расщепляет С5 и тем самым инициирует спонтанную сборку литического комплекса из поздних компонентов — от С5 до С9. Поскольку каждый активированный фермент расщепляет много молекул следующего профермента, каскад активации ранних компонентов действует как усилитель: каждая молекула, активированная в начале всей цепи, приводит к образованию множества литических комплексов.
Активация комплемента
Система комплемента работает как биохимический каскад реакций. Комплемент активируется тремя биохимическими путями: классическим, альтернативным и лектиновым путём. Все три пути активации производят разные варианты C3-конвертазы (белка, расщепляющего С3). Классический путь (он был открыт первым, но эволюционно является новым) требует антител для активации (специфический иммунный ответ, приобретённый иммунитет), в то время как альтернативный и лектиновый пути могут быть активизированы антигенами без присутствия антител (неспецифический иммунный ответ, врождённый иммунитет). Итог активации комплемента во всех трёх случаях одинаков: C3-конвертаза гидролизует СЗ, создавая C3a и C3b и вызывая каскад дальнейшего гидролиза элементов системы комплемента и событий активации. В классическом пути для активации С3-конвертазы необходимо образование комплекса С4bC2a. Этот комплекс образуется при расщеплении С2 и С4 С1-комплексом. С1-комплекс, в свою очередь, для активации должен связаться с иммуноглобулинами класса М или G. C3b связывается с поверхностью болезнетворных микроорганизмов, что приводит к большей «заинтересованности» фагоцитов к связанным с СЗb клеткам (опсонизация). C5a — важный хемоаттрактант, помогающий привлекать в район активации системы комплемента новые иммунные клетки. И C3a, и C5a имеют анафилотоксическую активность, непосредственно вызывая дегрануляцию тучных клеток (как следствие — выделение медиаторов воспаления). C5b начинает формирование мембраноатакующих комплексов (МАК), состоящим из C5b, C6, C7, C8 и полимерного C9. МАК — цитолитический конечный продукт активации системы комплемента. МАК формирует трансмембранный канал, вызывающий осмотический лизис клетки-мишени. Макрофаги поглощают помеченные системой комплемента болезнетворные микроорганизмы.
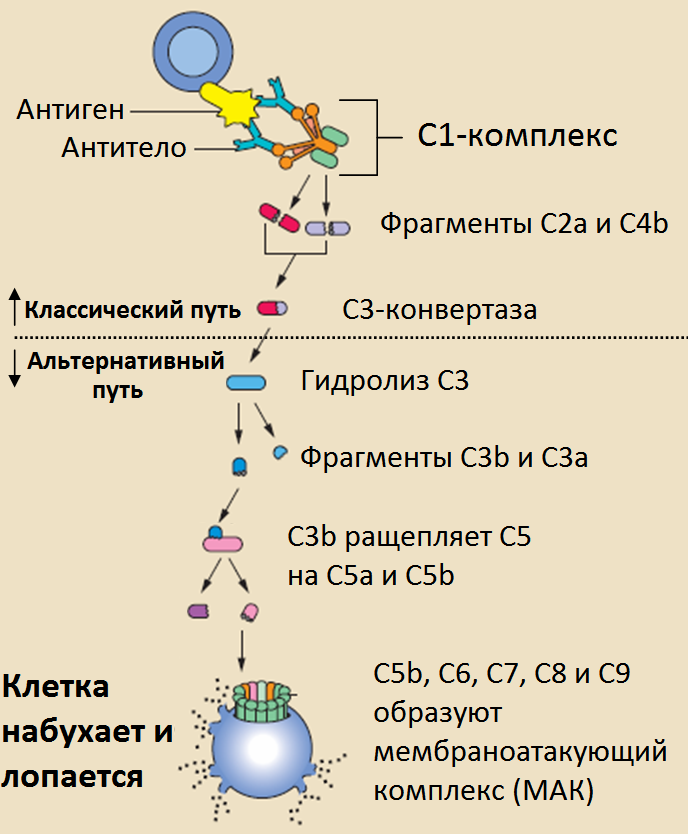
Биологические функции
Сейчас выделяют следующие функции:
- Опсонизирующая функция. Сразу вслед за активацией системы комплемента образуются опсонизирующие компоненты, которые покрывают патогенные организмы или иммунные комплексы, привлекая фагоциты. Наличие на поверхности фагоцитирующих клеток рецептора к С3b усиливает их прикрепление к опсонизированным бактериям и активирует процесс поглощения. Такое более тесное прикрепление С3b-связанных клеток или иммунных комплексов к фагоцитирующим клеткам получило название феномена иммунного прикрепления.
- Солюбилизация (то есть растворение) иммунных комплексов (молекулой C3b). При недостаточности комплемента развивается иммунокомплексная патология (СКВ-подобные состояния). [СКВ = системная красная волчанка]
- Участие в воспалительных реакциях. Активация системы комплемента приводит к выделению из тканевых базофилов (тучных клеток) и базофильных гранулоцитов крови биологически активных веществ (гистамина, серотонина, брадикинина), которые стимулируют воспалительную реакцию (медиаторов воспаления). Биологически активные компоненты, которые образуются при расщеплении С3 и С5, приводят к высвобождению вазоактивных аминов, таких как гистамин, из тканевых базофилов (тучных клеток) и базофильных гранулоцитов крови. В свою очередь это сопровождается расслаблением гладкой мускулатуры и сокращением клеток эндотелия капилляров, усилением сосудистой проницаемости. Фрагмент С5а и другие продукты активации комплемента содействуют хемотаксису, агрегации и дегрануляции нейтрофилов и образованию свободных радикалов кислорода. Введение С5а животным приводило к артериальной гипотонии, сужению легочных сосудов и повышению проницаемости сосудов из-за повреждения эндотелия.
Функции С3а:- выступать в роли хемотаксического фактора, вызывая миграцию нейтрофилов по направлению к месту его высвобождения;
- индуцировать прикрепление нейтрофилов к эндотелию сосудов и друг к другу;
- активировать нейтрофилы, вызывая в них развитие респираторного взрыва и дегрануляцию;
- стимулировать продукцию нейтрофилами лейкотриенов.
- Цитотоксическая, или литическая функция. В конечной стадии активации системы комплемента образуется мембраноатакующий комплекс (МАК) из поздних компонентов комплемента, который атакует мембрану бактериальной или любой другой клетки и разрушает её.
- Фактор С3е, образующийся при расщеплении фактора С3b, обладает способностью вызывать миграцию нейтрофилов из костного мозга, и в таком случае быть причиной лейкоцитоза.
Классический путь
Классический путь запускается активацией комплекса С1 (он включает одну молекулу С1q и по две молекулы С1r и С1s). Комплекс С1 связывается с помощью С1q с иммуноглобулинами классов М и G, связанными с антигенами. Гексамерный C1q по форме напоминает букет нераскрытых тюльпанов, «бутоны» которого могут связываться с Fc участком антител. Для инициации этого пути достаточно единственной молекулы IgM, активация молекулами IgG менее эффективна и требует больше молекул IgG.
С1q связывается прямо с поверхностью патогена, это ведет к конформационным изменениям молекулы С1q, и вызывает активацию двух молекул сериновых протеаз С1r. Они расщепляют С1s (тоже сериновую протеазу). Потом комплекс С1 связывается с С4 и С2 и затем расщепляет их, образуя С2а и С4b. С4b и С2а связываются друг с другом на поверхности патогена, и образуют С3-конвертазу классического пути, С4b2а. Появление С3-конвертазы приводит к расщеплению С3 на С3а и С3b. С3b образует вместе с С2а и С4b С5-конвертазу классического пути. С5 расщепляется на C5a и C5b. C5b остается на мембране и соединяется с комплексом C4b2a3b. Потом соединяются С6, С7, С8 и С9, которая полимеризуется и возникает трубочка внутри мембраны. Тем самым нарушается осмотический баланс и в результате тургора бактерия лопается. Классический путь действует более точно, поскольку так уничтожается любая чужеродная клетка.
Альтернативный путь
Альтернативный путь запускается гидролизом C3 прямо на поверхности патогена. В альтернативном пути участвуют факторы В и D. С их помощью происходит образование фермента СЗbBb. Стабилизирует его и обеспечивает его длительное функционирование белок P. Далее РС3bBb активирует С3, в результате образуется С5-конвертаза и запускается образование мембраноатакующего комплекса. Дальнейшая активация терминальных компонентов комплемента происходит так же, как и по классическому пути активации комплемента. В жидкости в комплексе СЗbBb В заменяется Н-фактором и под воздействием дезактивирующего соединения (Н) превращается в С3bi. Когда микробы попадают в организм, комплекс СЗbBb начинает накапливаться на мембране. Он соединяется с С5, который расщепляется на C5a и C5b. C5b остается на мембране. Потом соединяются С6, С7, С8 и С9. После соединения С9 с С8, происходит полимеризация С9 (до 18 молекул сшиваются друг с другом) и образуется трубочка, которая пронизывает мембрану бактерии, начинается закачка воды и бактерия лопается.
Альтернативный путь отличается от классического следующим: при активации системы комплемента не нужно образование иммунных комплексов, он происходит без участия первых компонентов комплемента — С1, С2, С4. Он также отличается тем, что срабатывает сразу же после появления антигенов — его активаторами могут быть бактериальные полисахариды и липополисахариды (являются митогенами), вирусные частицы, опухолевые клетки.
Лектиновый (маннозный) путь активации системы комплемента
Лектиновый путь гомологичен классическому пути активации системы комплемента. Он использует лектин, связывающий маннозу, (MBL) — белок, подобный C1q классического пути активации, который связывается с маннозными остатками и другими сахарами на мембране, что позволяет распознавать разнообразные болезнетворные микроорганизмы. MBL — сывороточный белок, принадлежащий к группе белков коллектинов, который синтезируется преимущественно в печени и может активировать каскад комплемента, непосредственно связываясь с поверхностью патогена.
В сыворотке крови MBL формирует комплекс с MASP-I и MASP-II (Mannan-binding lectin Associated Serine Protease, связывающие MBL сериновые протеазы). MASP-I и MASP-II весьма схожи с C1r и C1s классического пути активации и, возможно, имеют общего эволюционного предшественника. Когда несколько активных центров MBL связываются определенным образом c ориентированными маннозными остатками на фосфолипидном бислое болезнетворного микроорганизма, MASP-I и MASP-II активируются и расщепляют белок C4 на C4a и C4b, а белок С2 на C2a и C2b. Затем C4b и C2a объединяются на поверхности болезнетворного микроорганизма, формируя C3-конвертазу, а C4a и C2b действуют как хемоаттрактанты для клеток иммунной системы.
Регуляция системы комплемента
Система комплемента может быть очень опасной для тканей хозяина, поэтому её активация должна хорошо регулироваться. Большинство компонентов активны только в составе комплекса, при этом их активные формы способны существовать очень короткое время. Если в течение этого времени они не встретятся со следующим компонентом комплекса, то активные формы теряют связь с комплексом и становятся неактивными. Если концентрация какого-то из компонентов ниже пороговой (критической), то работа системы комплемента не приведет к физиологическим последствиям. Система комплемента регулируется специальными белками, которые находятся в плазме крови даже в большей концентрации, чем сами белки системы комплемента. Эти же белки представлены на мембранах собственных клеток организма, предохраняя их от атаки со стороны белков системы комплемента.
Регуляторные механизмы в основном действуют в трех точках.
- С1. Ингибитор С1 контролирует классический и лектиновый пути активации. Действует двумя путями: ограничивает действие С4 и С2 с помощью связывания C1r- и С1s-протеаз и подобным образом выключает лектиновый путь, удаляя ферменты MASP из MBP-комплекса.
- С3-конвертаза. Время жизни С3-конвертазы уменьшают факторы ускорения распада. Некоторые из них находятся на поверхности собственных клеток (например, DAF и CR1). Они действуют на С3-конвертазы и классического, и альтернативного путей активации. DAF ускоряет распад С3-конвертазы альтернативного пути. СR1 (C3b/C4b receptor) расположен главным образом на поверхности эритроцитов и отвечает за удаление из плазмы крови опсонизированных иммунных комплексов. Другие регуляторные белки производятся печенью и в неактивном состоянии растворены в плазме крови. Фактор I — сериновая протеаза, расщепляющая C3b и C4b. С4-связывающий белок (C4BP) расщепляет С4 и помогает фактору I расщеплять C4b.Фактор H связывается с гликозаминогликанами, которые есть на собственных клетках, но не на клетках патогенов. Этот белок является кофактором фактора I, а также ингибирует активность C3bBb.
- С9. CD59 и Гомологичный Фактор Ограничения ингибируют полимеризацию С9 во время образования мембраноатакующего комплекса, не давая ему сформироваться. Используется ВИЧ и цитомегаловирусом для защиты от системы комплемента организма хозяина.
Роль системы комплемента при болезнях
Система комплемента играет большую роль во многих болезнях, связанных с иммунитетом.
При болезнях иммунных комплексов комплемент провоцирует воспаление главным образом двумя путями:
- c C3b и C4b, фиксированными на иммунных комплексах, связываются лейкоциты, активируемые и привлекаемые в места отложения этих комплексов образовавшимися здесь анафилатоксинами. Так начинается повреждение тканей при синдроме Гудпасчера (системный капиллярит с преимущественным поражением легких и почек по типу геморрагических пневмонита и гломерулонефрита). Для подавления воспалительной реакции на экспериментальных моделях этого заболевания достаточно уменьшить содержание в крови комплемента или нейтрофилов.
- МАК (мембраноатакующий комплекс), внедряясь в мембрану собственных клеток организма, повреждает мембрану. При этом происходит высвобождение метаболитов арахидоновой кислоты — простагландинов. Этим обусловлено повреждение тканей при мембранозном нефрите, который в эксперименте удается вызвать антителами к субэпителиальным антигенам. Воспалительную реакцию в этом случае не подавляет устранение нейтрофилов, однако она полностью отсутствует у животных, дефицитных по C5.
Уже в первые часы после заражения геморрагической лихорадкой Эбола система комплемента блокируется

3.7. Анафилатоксины
Образующиеся в ходе комплементарного каскада фрагменты С3а, С4а, С5а, получили название анафилатоксины. Эти пептиды - гормоноподобные молекулы, взаимодействующие со своими специфическими рецепторами на поверхности различных клеток - полиморфноядерных лейкоцитов, моноцитов, макрофагов, тучных клеток, гладкомышечных клеток. Клеточные ответы на анафилатоксины включают: высвобождение гистамина, серотонина, гидролитических ферментов, фактора активации тромбоцитов, интерлейкина-1, активацию метаболизма арахидоновой кислоты, образование активных соC4bC2aC3b или C3bBbC3b соединений кислорода, хемотаксис лейкоцитов, сокращение клеток гладкой мускулатуры. С3а и С5а могут быть регуляторами иммунного ответа, - С3а подавляет, С5а стимулирует образование антител.
3.8. Эйкозаноиды
К медиаторам воспаления относятся эйкозаноиды – производные двадцати-(эйкоза-) углеродных полиненасыщенных жирных кислот. Наибольшее значение имеют продукты метаболизма арахидоновой кислоты, имеющей 20 углеродных атомов и 4 двойных связи (С20:4). Арахидоновая кислота входит в состав фосфолипидов клеточных мембран. Высвобождается из них под действием фосфолипаз, - либо фосфолипазы А2, либо действующих последовательно фосфолипазы С и диацилглицеридлипазы. Дальнейшие превращения свободной арахидоновой кислоты могут происходить различными путями. Наиболее изучены два пути: циклооксигеназный с образованием простагландинов и тромбоксана А2 – и липоксигеназный – с образованием лейкотриенов
3.9. NAMPT и NAPRT
Никотинамидфосфорибозилтрансфераза (NAMPT) и никотинатфосфорибозилтрансфераза (NAPRT) - это два внутриклеточных фермента, которые катализируют первую стадию биосинтеза НАД из никотинамида и никотиновой кислоты соответственно. Путем точной настройки внутриклеточных уровней НАД они участвуют в регуляции / перепрограммировании клеточного метаболизма и в контроле активности НАД-зависимых ферментов, включая сиртуины, PARP и НАДазы. Однако в ходе эволюции они оба приобрели новые функции в качестве внеклеточных эндогенных медиаторов воспаления. Хорошо известно, что клеточный стресс и / или повреждение вызывают высвобождение во внеклеточную среду эндогенных молекул, называемых аларминами или молекулярными паттернами, связанными с повреждениями (DAMP), которые модулируют иммунные функции посредством рецепторов распознавания паттернов связывания (PRR), таких как Toll -подобные рецепторы (TLR) и активируют воспалительные реакции. Все больше данных свидетельствует о том, что внеклеточные (е) NAMPT и eNAPRT являются новыми растворимыми факторами с цитокин / адипокином / DAMP-подобным действием. Повышенный eNAMPT был зарегистрирован при нескольких метаболических и воспалительных нарушениях, включая ожирение, диабет и рак, в то время как eNAPRT становится биомаркером сепсиса и септического шока.Одна из ключевых ролей врожденной иммунной системы - инициировать иммунный ответ против инвазивных патогенов. Патоген-ассоциированные молекулярные структуры (PAMP) включают сахара / липопротеины или нуклеиновые кислоты [то есть бактериальную ДНК в виде неметилированных повторов динуклеотидного CpG, двухцепочечной (ds) или одноцепочечной (ss) РНК] (1, 2). PAMP могут инициировать иммунные ответы через активацию классических рецепторов распознавания образов (PRR), среди которых есть toll-подобные рецепторы (TLR), NOD-подобные рецепторы (NLR), рецепторы, подобные индуцибельному гену I ретиноевой кислоты (RIG-I). (RLR), рецепторы лектина C-типа (CLR), множественные внутриклеточные ДНК-сенсоры и другие рецепторы DAMP, не относящиеся к PRR (2–4). Однако эти рецепторы могут быть задействованы также эндогенными лигандами. В настоящее время широко признано, что клетки в условиях гипоксии, ацидоза, окислительно-восстановительного дисбаланса, гипертонического / гипотонического стресса и нарушений внутриклеточных ионов или цитоскелета могут выделять небольшие эндогенные молекулы, называемые молекулярными паттернами, связанными с повреждениями (DAMP), или иногда «сигналами опасности» или «Alarmins», запускающие иммунные ответы через активацию PRR (4–6). Интересно, что многие из этих DAMPs обладают хорошо охарактеризованной внутриклеточной функцией и были случайно идентифицированы во внеклеточном пространстве, где они инициируют воспалительные реакции, независимо от патогенной инфекции, явление, называемое стерильным воспалением (4, 7, 8). Подобно воспалению, вызванному патогенами, DAMP могут праймировать нейтрофилы, макрофаги и дендритные клетки (DC), а также неиммунные клетки, включая эндотелиальные и эпителиальные клетки и фибробласты (7). Активация этих клеток приводит к выработке нескольких цитокинов и хемокинов, которые, в свою очередь, привлекают воспалительные элементы и запускают адаптивные иммунные ответы. Хотя стерильное воспаление играет важную роль в восстановлении и регенерации тканей, неразрешенное хроническое воспаление вредно для хозяина, приводя к развитию метаболических, нейродегенеративных, аутоиммунных нарушений и рака.
С момента их первоначального определения как DAMPs в 2003 году список эндогенных молекул значительно увеличился (4) и теперь включает высокоподвижный белок группы 1 (HMGB-1), белки теплового шока (HSP), гистоны и компоненты внеклеточного матрикса (для например, гиалуроновая кислота и бигликан). Все эти молекулы обладают провоспалительными функциями, связываясь с TLR. HMGB-1 - один из наиболее изученных DAMP. Это белок, связывающий ядерную ДНК, который можно найти во внеклеточном пространстве не только в результате некроза, но и с помощью специальных механизмов секреции (9, 10). Внеклеточно HMGB-1 вызывает провоспалительные эффекты, связанные с последующим связыванием TLR4 и активацией сигнального пути ядерного фактора каппа B (NF-kB) (11, 12). В моделях на животных HMGB-1 является поздним медиатором летального системного воспаления, участвующим в отсроченной летальности эндотоксина (13). Другие DAMP включают F-актин, белок 130, связанный с Sin3A (SAP130), β-глюкозилцерамид и N-гликаны, связывающиеся с CLR; кристаллы мононатрия урата (MSU), кристаллы холестерина, β-амилоид (Aβ) и аденозин-5'-трифосфат (АТФ), которые активируют инфламмасому NLRP3 (4). Кроме того, многочисленные цитокины [например, интерлейкин (IL) -1β, фактор некроза опухоли (TNF) и интерферон I типа (IFN-I)], провоспалительные белки, такие как индуцированный интерфероном белок 35, и биоактивные липиды, такие как лизофосфолипиды называются «индуцибельными DAMP» или «условными DAMP» (14).
Нуклеотиды и нуклеозиды, долгое время считавшиеся просто переносчиками электронов, участвующими в поддержании энергетического метаболизма, вызывают интерес вместе с сетью ферментов, контролирующих их синтез и разложение. Интересно, что хотя все эти факторы обладают хорошо охарактеризованной внутриклеточной функцией, они могут высвобождаться во внеклеточное пространство, где они связываются и активируют различные наборы клеточных рецепторов, включая пуринергические и PRR. Например, АТФ, ключевая молекула внутриклеточной энергии, может массово высвобождаться за счет пассивной утечки, когда клетки травмируются, подвергаются стрессу или даже некротизируются, действуя как DAMP (15). Внеклеточный АТФ и его производные нуклеотиды (аденозин, АМФ, АДФ), синтезируемые эндонуклеотидазами, достигают многих из своих эффектов через пуринергические рецепторы, через воспалительные каскады и продукцию провоспалительных цитокинов (16, 17). Среди ферментов, участвующих в синтезе никотинамидадениндинуклеотида (НАД), никотинамидфосфорибозилтрансфераза (NAMPT) - предмет этого обзора - выступает в качестве нового медиатора воспаления. Внутриклеточно он катализирует первую и лимитирующую стадию биосинтеза НАД из никотинамида (Nam) (18, 19). Сообщается о повышении уровня eNAMPT в условиях острого или хронического воспаления (18, 20-25). Эффекты eNAMPT в основном связаны с активацией воспалительной сигнатуры, в основном в макрофагах, с недавними данными, предполагающими, что он связывает TLR4, таким образом добавляя фермент к ряду «опасных» сигналов, активирующих этот рецептор (26). NAMPT структурно и функционально связан со вторым ферментом биосинтеза NAD (NBE), то есть никотинатфосфорибозилтрансферазой (NAPRT), который ограничивает скорость в пути восстановления NAD, который начинается с образования никотиновой кислоты (Na) (27–29). Наша группа недавно обнаружила присутствие NAPRT во внеклеточных жидкостях (eNAPRT), подчеркнув роль этого фермента в качестве лиганда для TLR4.
В этом обзоре суммируются текущие знания о NAMPT и NAPRT, как о внутриклеточных NBE, участвующих в регуляции / перепрограммировании клеточного метаболизма, и о цитокинах / DAMP во внеклеточной среде. Наконец, мы обсудим роль этих ферментов, особенно в отношении развития воспалительных состояний, включая рак, и их потенциальную терапевтическую ценность.
Уровни NAD модулируют клеточные транскрипционные ответы и метаболическую адаптацию
Наши знания о биологии НАД значительно выросли за последние несколько лет, в том числе о биосинтетических и деградирующих путях. Общее снижение клеточного НАД описано при многих возрастных заболеваниях, тогда как повышение уровня НАД связано с воспалительными состояниями, включая рак. На рисунке 1 показаны основные пути биосинтеза и потребления НАД, а также перекрестные помехи между внутриклеточным (i) НАД и eNAD.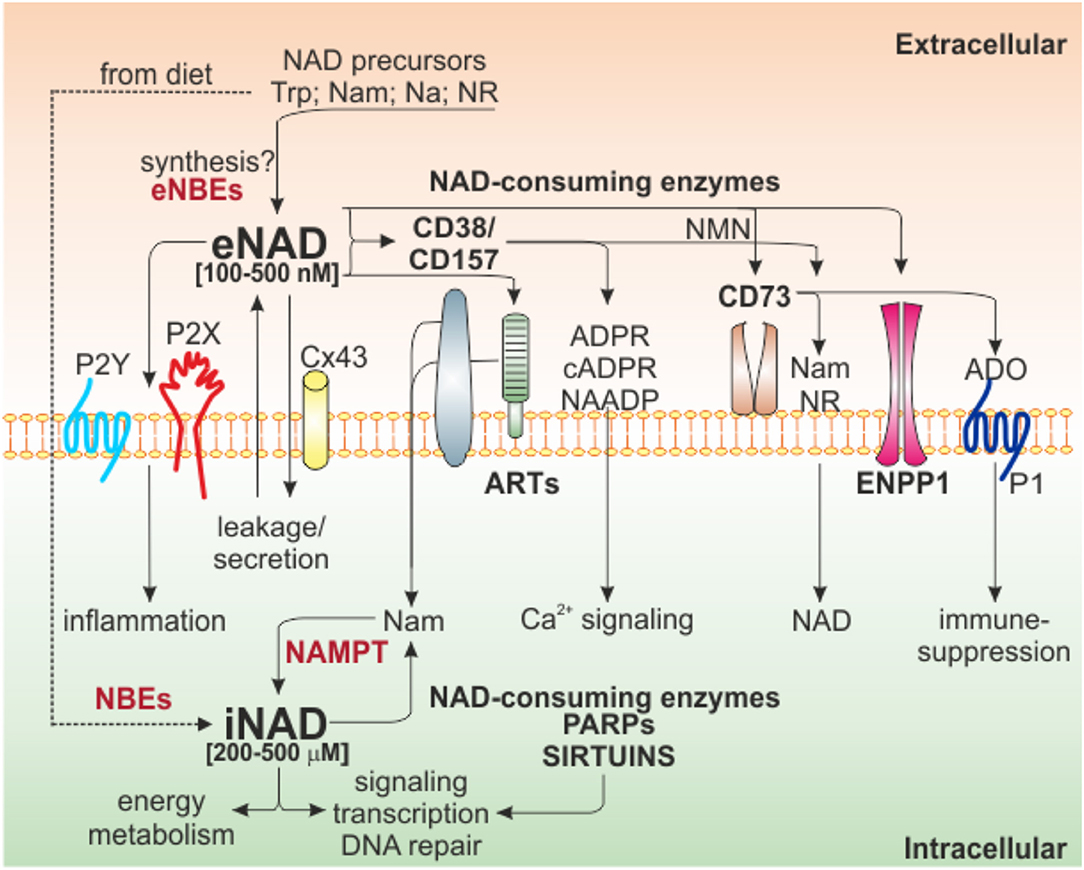
Рисунок 1. Интра/экстра клеточные взаимодействия НАД и ферменты, метаболизирующие НАД. Схематическое изображение сети NAD-метаболизирующих клеточных поверхностных и внутриклеточных ферментов и их продуктов. Некоторые предшественники НАД, полученные с пищей, могут быть интернализованы для генерации iNAD за счет активности NBE для поддержки энергетического метаболизма, передачи сигналов и других биологических процессов за счет активности внутриклеточных ферментов, потребляющих НАД (PARP и сиртуинов). Эти ферменты высвобождают Nam, который, в свою очередь, посредством NAMPT-зависимого пути восстановления поддерживает производство NAD. Попав во внеклеточное пространство из-за секреции / утечки через Cx43 или из-за прямого внеклеточного синтеза из предшественников (не подтверждено), eNAD может функционировать путем связывания пуринергических рецепторов (P2Y, P2X), что приводит к внутриклеточной передаче сигналов и воспалительным состояниям. . В качестве альтернативы, eNAD также может метаболизироваться серией эктоферментов на поверхности клетки (CD38 / CD157, ART, CD73, ENPP1), генерируя различные метаболиты (cADPR, ADPR и NAADP), участвующие в основном в передаче сигналов Ca2 +. Конечный продукт реакции, аденозин, может изменять передачу сигнала, воздействуя на пуринергические рецепторы P1, что обычно приводит к иммуносупрессии. В квадратных скобках указан диапазон iNAD [200–500 мкМ] или eNAD [100–500 нМ]. Trp, триптофан; Нам, никотинамид; NR, никотинамид рибозид; Na, никотиновая кислота; NBE, ферменты биосинтеза НАД; NAMPT, никотинамидфосфорибозилтрансфераза; АРТ, моноаденозиндифосфат (АДФ) -рибоза трансферазы; PARP, полимеразы поли-АДФ-рибозы; Сх43, коннексин 43; ADPR, ADP рибоза; cADPR, циклическая ADP рибоза; NAADP, адениндинуклеотидфосфат никотиновой кислоты; NMN, никотинамидмононуклеотид; АДО, аденозин; ENPP1, эктонуклеотидпирофосфатаза/фосфодиэстеразы.
NAD: энергетический кофактор
Как энергетический кофермент, НАД играет важную роль в качестве донора акцептора электронов в различных метаболических путях, включая цитозольный гликолиз, биосинтез серина, цикл трикарбоновых кислот (TCA), окислительное фосфорилирование, а также окислительно-восстановительные реакции гомеостаза окислительно-восстановительного состояния клетки (30, 31). Кофактор почти 300 дегидрогеназ, НАД в основном используется во время гликолиза на шестой стадии ферментативной цепи глицеральдегидфосфатдегидрогеназой (ГАФД) и в конце процесса лактатдегидрогеназой (ЛДГ), катализируя взаимное превращение пирувата и лактата и одновременно НАДН и НАД. Конечный гликолитический продукт пируват может метаболизироваться с образованием ацетилСоА комплексом пируватдегидрогеназы (PDC), реакция, сопровождающаяся восстановлением НАД до НАДН (32). Во время цикла TCA NAD восстанавливается до фрагментов NADH в несколько ключевых этапов под действием изоцитратдегидрогеназы (IDH), оксоглутаратдегидрогеназы (OGD) и малатдегидрогеназы (MDH). НАДН, образующийся во всех этих реакциях, работая как перераспределитель электронных эквивалентов, используется цепью переноса электронов (ETC) для генерации АТФ (33).Соотношение между НАД / НАДН и их относительной фосфорилированной формой (НАДФ / НАДФН) также имеет решающее значение для систем ферментативной защиты от окислительного стресса, регулируя окислительно-восстановительный гомеостаз через основные клеточные системы очистки, которыми являются глутатион (GSH / GSSG) и тиоредоксин. опосредованные (Trx-SH / Trx-SS) механизмы (34–37). В этом контексте НАДФН является незаменимым восстанавливающим агентом для элиминации АФК и окислительно-восстановительного гомеостаза, в первую очередь продуцируемого глюкозо-6-фосфатдегидрогеназой (G6PD) и -фосфоглюконатдегидрогеназой (6PGD) в пентозофосфатном пути (36).
НАД: Плейотропная сигнальная молекула
Независимо от его окислительно-восстановительных свойств, НАД также является субстратом ферментов, играющих фундаментальную роль в экспрессии генов и передаче сигналов в клетке (38). В этих реакциях NAD расщепляется по гликозидной связи между Nam и ADP-рибозой, приобретая характеристики сигнальной молекулы (27).nnБольшое семейство энзимов, потребляющих НАД, включает моноаденозиндифосфат (АДФ) -рибозо-трансферазы (АРТ) и поли-АДФ-рибозные полимеразы (ПАРП), НАД-зависимые деацетилазы, сиртуины (SIRT1-7) и циклические АДФ-рибозогидролазы. , NAD-гликогидролазы, эктонуклеотидпирофосфатаза / фосфодиэстеразы и экто-5'-нуклеотидаза (CD38 / CD157 и ENPP1 / CD73) (19, 39, 40) (Рисунок 1).nnБлагодаря своей функциональной активности посттрансляционных модификаций (АДФ-рибозилирование и деацетилирование) или посредством модуляции передачи сигналов Ca2 +, эти ферменты регулируют транскрипцию генов, дифференциацию клеток, развитие клеточного цикла, циркадный ритм, восстановление ДНК, стабильность хроматина, адаптацию клеток к сигналы стресса и иммунные ответы (41, 42). Следовательно, PARP и сиртуины представляют собой связующие элементы между метаболическим состоянием клетки и ее сигнальной и транскрипционной активностью (43).nnВнеклеточный НАД и его биологическая рольnКонцентрация eNAD находится в диапазоне 100–500 нМ, что значительно ниже, чем его внутриклеточные уровни (200–500 мкМ) (39, 44–46). Уровни eNAD и iNAD тесно связаны из-за внутримембранного транспорта предшественников NAD, промежуточных продуктов реакции и самого NAD (47). Каноническая точка зрения состоит в том, что NAD неспособен пересекать липидные бислои, но он входит в клетку с помощью специализированных транспортеров NAD, таких как каналы коннексина 43 (Cx43), или выходит через экзоцитоз (45, 48-50). Кроме того, стрессовые условия окружающей среды могут вызывать высвобождение НАД (51–53). С другой стороны, существует ли прямой синтез eNAD, остается спорным (39) (Рисунок 1), несмотря на присутствие внеклеточных предшественников и биосинтетических ферментов. В частности, известно, что среди различных форм витамина B3 (предшественник NAD) транспорт Na опосредуется системами мембранных носителей, потенциально включая pH-зависимый анионный антипортер или котранспортер протонов (54, 55). Nam присутствует внеклеточно, и его поглощение возможно либо в виде прямого транспорта в интактной форме, либо в виде метаболитов спасательного пути. Однако было показано, что субстраты NAMPT АТФ и 5-фосфорибозил-1-пирофосфат (PRPP) недоступны в достаточных количествах во внеклеточном пространстве (56), чтобы поддерживать прямую генерацию eNAD.nneNAD может связывать различные подтипы пуринергических рецепторов P2, включая P2Y11, что приводит к открытию каналов Ca2 + L-типа и активации сигнального каскада cAMP / cADPR / [Ca2 +] i, что в конечном итоге вызывает усиление пролиферации и миграции (57). В Т-клетках и моноцитах активация рецептора P2X7 обычно приводит к интернализации Ca2 +, открывая неселективные большие мембранные поры, вызывая гибель клеток (58, 59). eNAD также действует как нейротрансмиттер, высвобождаемый стимулированными окончаниями нейронов центральной нервной системы и периферической нервной системы млекопитающих, связываясь с постсинаптическими рецепторами P2Y1, подобно АТФ (60).nnОчень низкие уровни eNAD обусловлены его быстрым метаболизмом / деградацией NAD-катаболическими ферментами, присутствующими на поверхности клетки (61), что позволяет предположить, что метаболиты NAD также могут опосредовать клеточные ответы во внеклеточной среде.nneNAD расщепляется тремя основными классами специфических эктоферментов: CD38 и CD157 (62, 63), ART (64), ENPP1 и CD73 (61, 65, 66). NADase, ENPP1 и CD73 могут приводить к образованию аденозина (ADO), мощного иммунодепрессанта, независимо от активности CD39 (61, 67, 68). Помимо образования ADO, eNAD может расщепляться до никотинамидмононуклеотида (NMN) с помощью CD38, генерируя Nam, который может проникать через плазматические мембраны и повторно превращаться в NAD через NAMPT и NMN-аденилилтрансферазу (NMNAT) (69). С другой стороны, NMN также может использоваться CD73, который генерирует никотинамид рибозид (NR) (66, 70), который, вероятно, через уравновешивающие переносчики нуклеозидов (ENT), может быть импортирован как предшественник NAD (71, 72) (рис. 1). Недавно Slc12a8 был идентифицирован как специфический переносчик NMN (73), предполагая, что NMN может быть интернализован без преобразования в NR. Исследования паттерна экспрессии клеточного типа этого переносчика прояснят эту возможность.
Биосинтез NAD: ферментативные функции NAMPT и NAPRT
Оборот NAD в клетке является динамическим, демонстрируя циркадные колебания, которые регулируются механизмом основных часов CLOCK: BMAL1 (74, 75). Общие внутриклеточные уровни поддерживаются между 200 и 500 мкМ, в зависимости от типа клетки или ткани, увеличиваясь в ответ на различные стимулы (43). Гомеостаз НАД является результатом баланса между рядом реакций, потребляющих НАД, и путями биосинтеза НАД через три различных пути: путь биосинтеза de novo, путь Прейсс-Хэндлера и путь спасения, как описано в Houtkooper et al. . (27), Ruggieri et al. (29) и Audrito et al. (42) и показано на рисунке 2.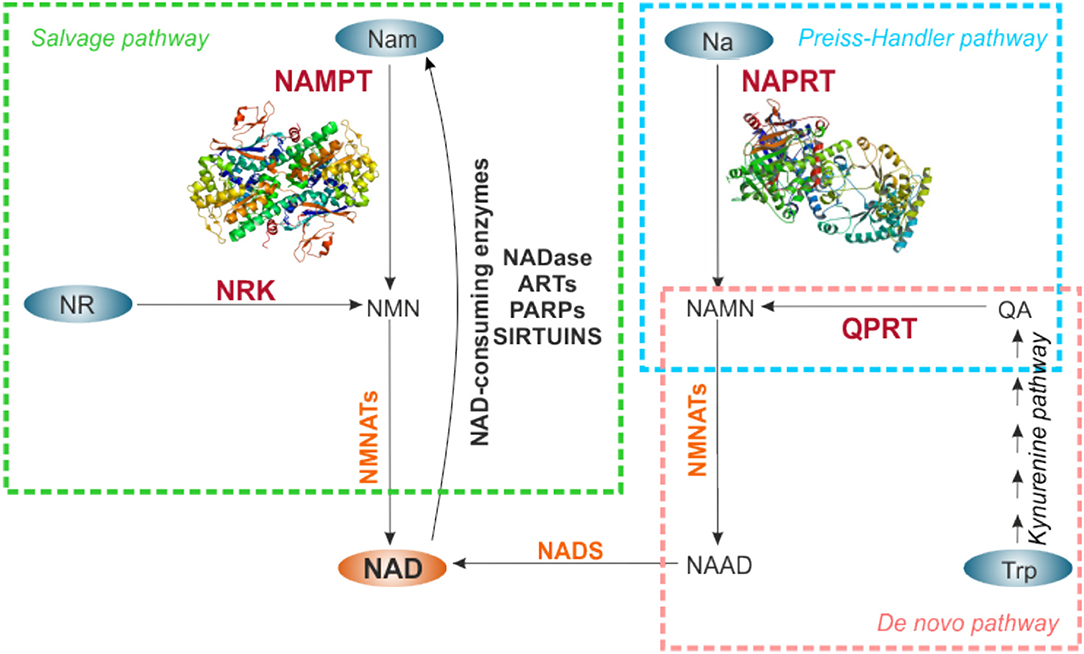
Рисунок 2. Пути биосинтеза НАД. НАД можно синтезировать de novo, начиная с Trp (розовый прямоугольник), или через пути спасения от Nam и NR (зеленый прямоугольник), или метаболизируя Na по пути Прейсс-Хэндлера (голубой прямоугольник). Предшественники НАД обозначены синими овалами. Ферменты, ограничивающие скорость каждого пути биосинтеза, обозначены красным цветом, другие ферменты, участвующие в реакциях, - оранжевым. Для NAMPT и NAPRT показаны кристаллические структуры. НАД, синтезируемый из Nam с помощью NAMPT, в свою очередь, используется потребляющими NAD активностями ферментов, которые высвобождают Nam, делая его доступным для непрерывной регенерации NAD. NAMPT, никотинамидфосфорибозилтрансфераза; НАПРТ, никотинатфосфорибозилтрансфераза; NRK, никотинамид-рибозидкиназа; QPRT, хинолинатфосфорибозилтрансфераза; NMNAT, никотинамидмононуклеотид аденилтрансферазы; НАДС, НАД-синтаза; Нам, никотинамид; NR, никотинамид рибозид; Na, никотиновая кислота; Trp, триптофан; QA, хинолиновая кислота; NMN, никотинамидмононуклеотид; НАМН, никотинатмононуклеотид; НААД, никотинатадениндинуклеотид; НАДаза, НАД-гликогидролаза; АРТ, моноаденозиндифосфат (АДФ) -рибоза трансферазы; PARP, полимеразы поли-АДФ-рибозы.
В частности, de novo биосинтез НАД начинается с катаболизма аминокислоты триптофана до кинуренина индоламин-2,3-диоксигеназой. Затем кинуренин метаболизируется посредством кинуренинового пути до хинолиновой кислоты (QA), которая превращается хинолатфосфорибозилтрансферазой (QPRT), ограничивающим скорость ферментом, в мононуклеотид натрия (NaMN). Путь Прейсса-Хэндлера метаболизирует NaMN, полученный из кинуренинового пути, или NaMN, полученный из пищи, или Na, как продукт дезамидирования Nam кишечной флорой (76) до NAD, посредством NAPRT-ограничивающей активности. В пути спасения NAMPT метаболизирует Nam и PRPP до NMN на этапе ограничения скорости, который затем превращается в NAD. В другом способе спасения NR, полученный из диеты, может использоваться никотинамид-рибозидкиназой (NRK) для генерации NAD (рис. 2).
Количественно путь спасения Nam является наиболее значимым в клетках млекопитающих. Это наблюдение подтверждается несколькими доказательствами. Во-первых, Nam является наиболее распространенным предшественником НАД в кровотоке (39), и его можно легко ввести с пищей (витамин B3). Во-вторых, Nam является побочным продуктом активности всех ферментов, метаболизирующих НАД, что увеличивает его доступность (77). В-третьих, ограничивающий скорость фермент NAMPT (EC 2.4.2.12) экспрессируется во всех тканях млекопитающих (78), как подробно описано ниже. Связанная с этим делеция гена NAMPT у мышей является эмбрионально летальной (79), что свидетельствует о важности этого пути для регенерации NAD. На этом пути Nam N-methyltransefase (NNMT) недавно стала эволюционно консервативным регулятором доступности Nam. Фактически, NNMT N-метилирует Nam, предотвращая его накопление и ингибирование ферментов, потребляющих NAD, а с другой стороны, ограничивая его доступность для NAMPT (80, 81).
Функциональный NAMPT образует гомодимер, катализирующий превращение Nam и PRPP в NMN. Исследования структурного и сайт-направленного мутагенеза Khan et al. продемонстрировали, что Asp219 играет фундаментальную роль в определении субстратной специфичности NAMPT (82). Wang et al. показали, что NAMPT обладает аутофосфорилирующей активностью и гидролизует АТФ. Аутофосфорилирование может повысить его ферментативную активность (83). Недавно было обнаружено, что NAMPT является прямым субстратом деацетилирования SIRT6, посттрансляционного механизма, который регулирует его ферментативную активность (84). Напротив, мутации His247, центрального консервативного остатка в активном центре фермента, значительно снижают или отменяют ферментативную активность NAMPT (83).
NAPRT (EC 2.4.2.11) катализирует превращение Na и PRPP в NaMN и пирофосфат (PPi). Фермент, первоначально названный пирофосфорилаза NaMN, был впервые описан Хэндлером в человеческих эритроцитах, где он увеличивает уровни НАД (85).
Активность NAPRT более тканеспецифична. Хотя активность фермента может быть обнаружена в большинстве тканей мыши (86), Na действует как более эффективный предшественник, чем Nam, в печени, кишечнике, сердце и почках мышей (87). Кроме того, Na более эффективен, чем Nam, в повышении уровня НАД в клетках, подверженных окислительному стрессу (56, 85, 88).
В отличие от NAMPT, NAPRT не ингибируется NAD, что объясняет его значительно более высокую эффективность в повышении уровня NAD in vivo (56, 89). Более того, NAPRT сильно активируется фосфатом (85), в то время как АТФ ведет себя как аллостерический модулятор фермента (29, 85, 90).
В 2015 году Марлетта и др. разрешили структуру человеческого (h) NAPRT, подчеркнув высокую степень структурной гомологии между человеческими и бактериальными NaPRTases из-за эволюционной адаптации (91). Как и в случае с NAMPT, функциональный фермент NAPRT работает как димеры, и, несмотря на очень ограниченное сходство последовательностей, hNAPRT демонстрирует молекулярную складку, которая очень похожа на ту, что впервые была описана для hNAMPT (83). Это открыло новые гипотезы об общих мотивах в NAMPT и NAPRT, участвующих в связывании внеклеточных белков с рецептором, как описано в разделе Функции eNAMPT.
Наличие этих множественных путей биосинтеза NAD, скорее всего, отражает различия в тканевом распределении и / или внутриклеточной компартментализации NBE (39, 46, 76, 92, 93). Наша группа недавно показала, что NAMPT и NAPRT в основном расположены в цитоплазме и ядре, тогда как NRK больше экспрессируется в митохондриях, влияя на уровни iNAD и ответ на ингибиторы NAMPT (39, 46, 76, 92, 93).
https://www.frontiersin.org/articles/10.3389/fonc.2020.00358/full
Front. Oncol., 19 March 2020 | https://doi.org/10.3389/fonc.2020.00358
4. Инфламмасома
Инфламмасо́ма (англ. inflammasome от англ. inflammation — воспаление) — многобелковый олигомерный комплекс, отвечающий за активацию воспалительного ответа. Инфламмасома способствует созреванию и секреции провоспалительных цитокинов интерлейкина-1β (IL-1β) и интерлейкина 18 (IL-18). Секреция этих цитокинов вызывает пироптоз — особый вид программируемой клеточной гибели. Нарушения в функционировании инфламмасом приводят к разнообразным болезням.
Инфламмасомы образуются в миелоидных клетках и являются частью врождённого иммунитета. В состав инфламмасомы могут входить такие белки, как каспаза 1, PYCARD, NLRPepeat-containing receptors, NLRs) и AIM2-подобные рецепторы (англ. AIM2-like receptors, ALRs). Состав конкретной инфламмасомы зависит от активатора, который запустил её образование. Так, состав инфламмасом, формирование которых активировала двуцепочечная РНК (дцРНК), отличается от такового у инфламмасом, сформированных под действием асбеста.
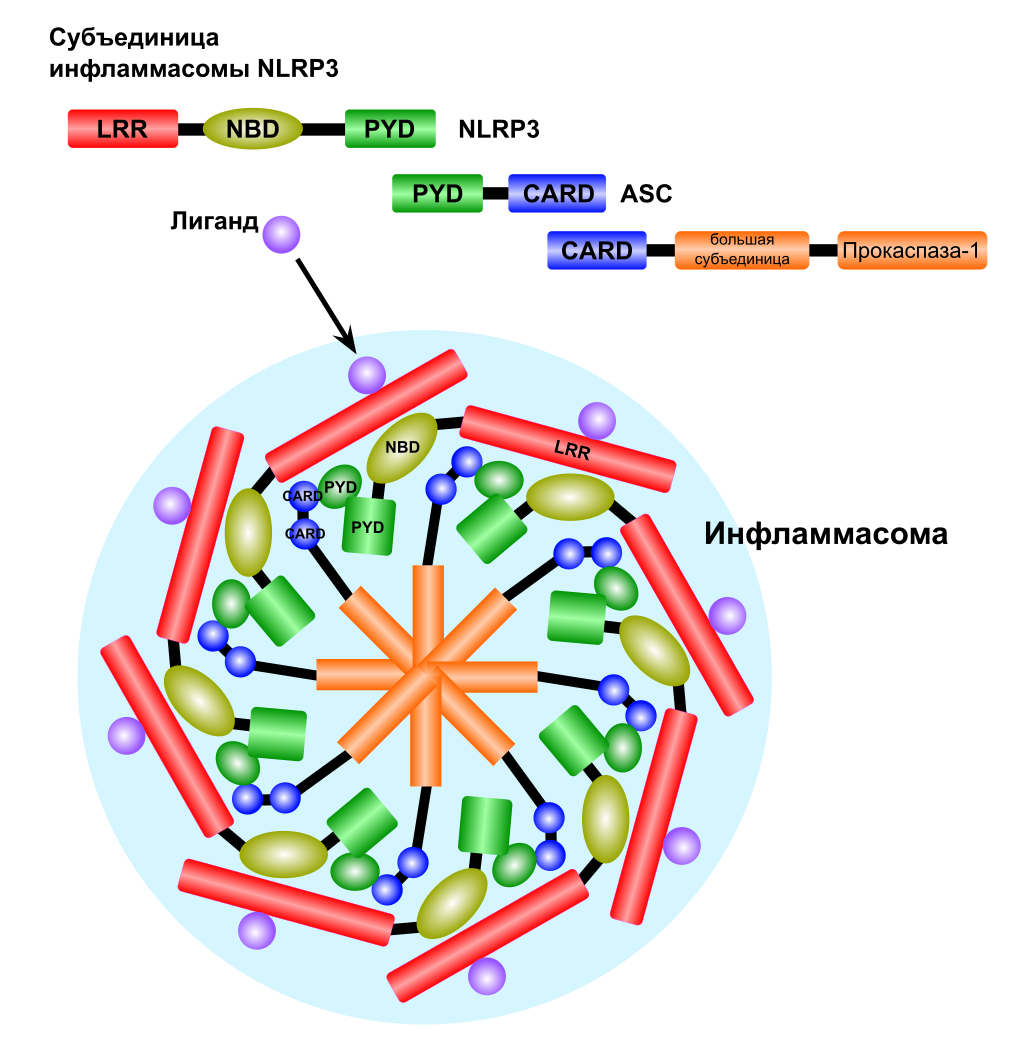
4.1. История
Инфламмасомы были описаны исследовательской группой под руководством Юрга Чоппа в 2002 году в Университете Лозанны. Исследователи смогли точно установить роль инфламмасом в развитии таких заболеваний, как подагра и сахарный диабет 2-го типа. Они обнаружили, что формирование инфламмасом могут запустить разнообразные сигналы опасности: вирусная ДНК, мурамилдипептид[en], асбест и кремний. Они также установили связь между метаболическим синдромом и инфламмасомами вида NLRP3. Когда они изучали NLRP3, то им удалось показать, что, когда инфламмасомы NLRP3 подавлены, то проявляется иммуносупрессивный эффект интерферона I типа[en]. Наконец, группа Чоппа запустила исследования и поиск лечения для многих заболеваний, связанных с инфламмасомами
4.2. Функции
Один из первых защитных механизмов, включающихся при инфекции, — это врождённый иммунитет, а именно, рецепторы опознавания паттерна, которые распознают особые молекулы (паттерны) на поверхности патогенов. Рецепторы опознавания паттерна могут располагаться как на мембранах клеток, как Toll-подобные рецепторы (TLRs) и рецепторы лектинов C-типа (CLRs), так и в цитоплазме, как Nod-подобные рецепторы (NLRs) и RIG-I-подобные рецепторы (RLRs). В 2002 году Юрг Чопп и его коллеги впервые сообщили, что подвид NLRs, известный как NLRP1, могут олигомеризоваться и собираться в комплекс, который активирует каскад каспазы 1, приводящий, в конце концов, к образованию провоспалительных цитокинов, особенно IL-1β и IL-18. Комплекс, формируемый NLRP1, был назван инфламмасомой[9]. Впоследствии были описаны другие виды инфламмасом, такие как NLRP3 и NLRC4. В 2009 году было описано новое семейство инфламмасом, содержащих белок AIM2, которые активируются в ответ на появление в цитоплазме клетки чужеродной двуцепочечной ДНК (дцДНК)
4.3. Нарушения
Подобно апоптосоме, запускающей гибель клетки по пути апоптоза, инфламмасома запускает воспалительный сигнальный каскад, приводящий к пироптозу — другой форме программируемой клеточной гибели. Активная инфламмасома связывается с прокаспазой-1 (предшественницей каспазы-1) посредством либо собственного домена привлечения и активации каспаз (CARD-домена от англ. caspase activation and recruitment domain) или через CARD-домен адаптерного белка[en] PYCARD, который связывается с инфламмасомой на этапе её формирования. Одна инфламмасома связывается с несколькими молекулами прокаспазы-1 (p45), запуская их автокаталитическое разрезание на две молекулы — p10 и p20. Эти две молекулы формируют гетеродимер, а два гетеродимера, связываясь друг с другом, образуют активную каспазу-1, которая инициирует несколько связанных с воспалением процессов, таких как созревание IL-1β и IL-18 из молекул-предшественников. Эти интерлейкины, в свою очередь, индуцируют секрецию интерферона γ и активируют натуральные киллеры. Далее происходит разрезание и инактивация интерлейкина-33 (IL-33), фрагментация ДНК[en] и формирование пор в клетке, подавление ферментов гликолиза, активация биосинтеза липидов и секреция молекул, способствующих восстановлению тканей, таких как предшественник интерлейкина-1α (IL-1α).
Показано что основной эндогенный продукт перекисного окисления липидов, 4-гидроксиноненаль, напрямую связывается с NLRP3 и ингибирует активацию воспаления вызываемого инфламмасомой NLRP3 независимо от передачи сигналов Nrf2 (Nuclear factor erythroid 2-related factor 2) и NF-κB
5. Пироптоз
Пиропто́з (англ. pyroptosis) — вид программируемой некротической гибели клетки, при котором в результате активации каспазы 1 происходит нарушение целостности плазматической мембраны и быстрое высвобождение наружу содержимого клетки. Характерной чертой пироптоза является зависимое от каспазы 1 активное выделение клеткой интерлейкинов IL‑1β и IL‑18, что приводит к воспалению. Пироптоз служит защитным механизмом врождённого иммунитета, ограничивающим размножение внутриклеточных патогенов, однако этот тип гибели клеток не ограничивается бактериальными инфекциями.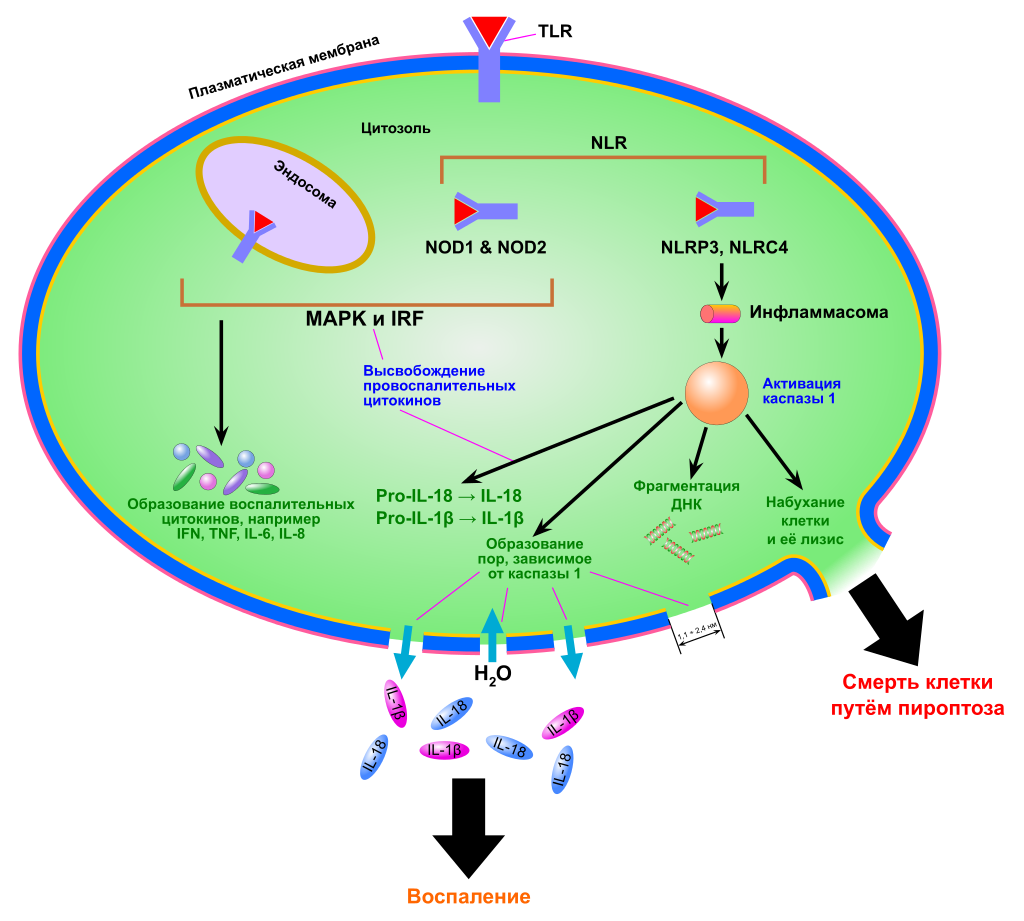
Впервые явление пироптоза описали Артуро Зихлински с коллегами в 1992 году, когда изучали литическую форму гибели макрофагов, заражённых Shigella flexneri. Поначалу этот вид клеточной гибели приняли за апоптоз, поскольку для него оказались характерны некоторые черты, присущие апоптозу: фрагментация ДНК, конденсация ядер, а также зависимость от каспаз. Позднее была описана форма клеточной гибели макрофагов, заражённых Salmonella enterica subsp. enterica (серовар Typhimurium); она оказалась зависимой от каспазы 1 и совершенно отличной от апоптоза. Для неё в 2001 году был предложен термин «пироптоз». Греческий корень «пиро» означает «огонь», что указывает на воспалительную природу этой формы гибели, а корень «птоз» («опадение») используется в названиях других форм клеточной гибели
5.1. Ключевые особенности
Пироптоз определяется рядом критериев. Во-первых, пироптоз запрограммирован воспалительными каспазами. Для пироптоза необходима протеолитическая активность каспаз, но не аутопротеолитический процессинг (то есть разрезание каспазами друг друга, приводящее к их активации). В большинстве случаев пироптоза каспазы активируются иначе, то есть не разрезанием друг друга, а при непосредственном участии особого комплекса — инфламмасомы, содержащей домен CARD. Во-вторых, активация воспалительных каспаз приводит к образованию пор в плазматической мембране, и клетка при этом становится проницаемой для красителей с малой молекулярной массой, для которых мембрана непроницаема, как то: 7-аминоактиномицин D[en] (7-AAD), бромистый этидий (EtBr) и иодистый пропидий[en] (PI). По-видимому, эти поры имеют диаметр 1,1—2,4 нм. При апоптозе, наоборот, клеточная мембрана остаётся интактной и клетка распадается на апоптотические тельца, которые не окрашиваются 7-AAD или PI.
После нарушения целостности мембраны в клетку устремляются ионы и вода, в результате чего клетка набухает и подвергается лизису, высвобождая наружу своё содержимое. После разрушения мембраны внутренняя сторона плазматической мембраны оказывается выставленной во внеклеточную жидкость, а потому может быть окрашена аннексином V[en], который связывается с фосфатидилсерином (PS) — фосфолипидом, встречающимся только во внутренней стороне клеточной мембраны. При апоптозе флиппаза перемещает PS в наружный слой мембраны, благодаря чему становится возможным окрашивание аннексином V. Таким образом, окрашивание аннексином V не даёт возможности отличить апоптоз от пироптоза.
В-третьих, при пироптозе в клетках происходит фрагментация ДНК[en], а TUNEL-анализ[en] даёт положительный результат, однако не такой интенсивный, как при апоптозе. Более того, «эффект лестницы» ДНК[en] (англ. DNA laddering) при пироптозе выражен слабо. Как и при апоптозе, происходит конденсация хроматина, однако ядро остаётся интактным. При апоптозе повреждение ДНК обеспечивается работой ДНКазы, активируемой каспазами[en] (CAD), а при пироптозе этот фермент остаётся связанным со своим ингибитором ICAD[en] (хотя в условиях in vitro каспаза 1 может расщеплять ICAD). Вместе с тем разрушение ДНК не является необходимым для пироптоза, и подавление фрагментации ДНК с помощью ингибиторов нуклеаз не предотвращает лизис клеток.
Наконец, при непрограммируемой некротической литической гибели клетки повреждение ДНК активирует полимеразу ADP-рибозы (PARP), которая расходует NAD+, тем самым понижая уровень АТР в клетке. Эффекторные каспазы апоптоза разрезают и инактивируют PARP, поддерживая уровень АТР в клетке, достаточный для дальнейшего хода апоптоза. Однако при пироптозе, индуцируемом Salmonella typhimurium, PARP не инактивируется, хотя in vitro каспаза 1 может разрезать PARP. Неизвестно, может ли каспаза 11 расщеплять ICAD или PARP in vitro, подобно каспазе 1. Макрофаги, лишённые PARP, успешно подвергались пироптозу, поэтому PARP не необходима для пироптоза. Было показано, что in vitro PARP-1 может выступать кофактором ядерного фактора-κВ (NF-κB) в регуляции транскрипции каспазы 11, индуцированной липополисахаридом (LPS). Однако нокдаун PARP-1 при помощи shРНК никак не влиял на индуцированную интерфероном γ (IFN-γ) транскрипцию каспазы 11, поэтому PARP-1, видимо, не участвует в пироптозе, опосредованном каспазой 11. Пироптоз отличен также и от программируемой некротической гибели клетки (некроптоза); ключевые отличия пироптоза, апоптоза и некроптоза резюмированы в таблице ниже
| Параметр сравнения | Пироптоз | Апоптоз | Некроптоз |
|---|---|---|---|
| Воспаление | Есть | Нет | Есть |
| Литический/нелитический | Литический | Нелитический | Литический |
| Инициаторные каспазы | Каспазы 1, 4, 5, 11 | Каспазы 2, 8, 9, 10 | Нет |
| Эффекторные каспазы | Нет | Каспазы 3, 6, 7 | Нет |
| Повреждение ДНК Эффект лестницы | Нет | Есть | Нет |
| TUNEL-анализ | Положительный | Положительный | Положительный |
| Разрушение ICAD | Нет | Да | Нет |
| Конденсация хроматина | Да | Да | Нет |
| Интактность ядра | Да | Нет | Да |
| Образование пор в мембране | Да | Нет | Да |
| Разрушение PARP | Нет | Да | Нет[4] |
| Окрашивание аннексином V | Положительное | Положительное | Положительное |
5.2. Молекулярные механизмы
Пироптоз запускается сигналами опасности, которые распознаются во вне- и внутриклеточной среде двумя группами рецепторов опознавания паттерна: Nod-подобными рецепторами (NLR), находящимися в цитоплазме, и Toll-подобными рецепторами (TLR), расположенными в плазматической мембране. Эти сигналы опасности могут выделяться патогенными организмами, при повреждениях тканей. При связывании с NLR внутриклеточных бактериальных, вирусных или принадлежащих самой клетке сигналов опасности начинается сборка мультибелкового комплекса — инфламмасомы (от англ. inflammation — воспаление). Сборка инфламмасомы приводит к активации каспазы 1, которая необходима для образования и выделения провоспалительных цитокинов. Наиболее хорошо изученная инфламмасома, NLRP3, имеет домены трёх главных типов: домены, содержащие обогащённые лейцином повторы (LRR), центральный нуклеотид-связывающий домен олигомеризации (NBD) и N-концевой пириновый домен (PYD). Взаимодействие каспазы 1 и NLRP3 осуществляется при помощи адаптерного белка ASC. ASC содержит домен активации и рекрутирования каспаз (CARD-домен), который связывается с прокаспазой 1 и облегчает её активацию через взаимодействие с её доменом CARD[9]. В результате прокаспазы 1 сближаются, димеризуются и разрезают друг друга на фрагменты p10 и p20, которые способы осуществлять процессинг про-IL-1β и про-IL-18. В других случаях инфламмасома активирует каспазу 1 через другие белки, содержащие домен CARD или PYD[10].
Ключевая роль каспазы 1 в пироптозе была продемонстрирована в 1995 году в экспериментах с мышами[en]*, нокаутными по каспазе 1: их клетки были неспособны к пироптозу. Впрочем, в 2011 году было показано, что все эти мыши несли ещё и сопутствующую мутацию в гене каспазы 11[en]. Поэтому каспаза 11 (человеческие ортологи — каспазы 4 и 5 также играет важную роль в пироптозе и врождённой защите от внутриклеточных патогенов. Более того, каспаза 11 может усиливать активацию каспазы 1 при инфекциях, вызванных грамотрицательными бактериями, так что обе эти каспазы могут инициировать пироптоз.
Для прохождения пироптоза необходима протеолитическая активность каспаз, и при пироптозе происходит разрушение или нарушение работы ряда белков, необходимых для функционирования и выживания клетки. К настоящему моменту известно множество мишеней каспазы 1. Среди них белки цитоскелета, внутриклеточного транспорта, трансляции и ключевых метаболических путей (например, гликолитические ферменты альдолаза, триозофосфатизомераза[en], глицеральдегид-3-фосфатдегидрогеназа[en], енолаза и пируваткиназа). Кроме того, каспаза 1 процессирует и другие каспазы: 4, 5 и 7. Хотя каспаза 7 является эффекторной каспазой апоптоза, её активация для пироптоза необязательна, что свидетельствует о различиях в сигнальных путях этих двух видов клеточной гибели.
Ранним этапом пироптоза является образование небольших пор в клеточной мембране, проницаемых для катионов. Это нарушает ионный баланс клетки и приводит к её набуханию и лизису. Через эти поры в клетку входят ионы Ca2+, которые участвуют во многих клеточных событиях, связанных с каспазой 1. Лизису клетки предшествуют конденсация ядра, фрагментация ДНК[en], а также секреция IL-1β и IL-18. Кальций также способствует экзоцитозу лизосом, которые «закрывают» прорехи в мембране и делают возможным высвобождение антимикробных соединений. Эти соединения убивают бактерий, находящихся во внутриклеточной среде. Кроме того, путём экзоцитоза до лизиса клетки наружу выделяются фагоцитированные частицы и внутриклеточные патогены.
В ходе пироптоза из клетки высвобождаются так называемые молекулярные паттерны, связанные с повреждениями (англ. Damage-associated molecular patterns, DAMP), которые во внеклеточной среде выступают сильными провоспалительными факторами. К их числу принадлежат АТФ, ДНК, РНК, белки теплового шока. DAMP запускают воспалительные процессы в клетках, активируют выделение цитокинов и усиливают нацеливание Т-клеток на определённые мишени. Один из DAMP, HMGB1, играет особую роль в пироптозе. HMGB1 — это ядерный транскрипционный фактор, который высвобождается наружу при пироптозе и активирует клеточные рецепторы TLR4 и RAGE, которые, в свою очередь, активируют выделение цитокинов и миграцию клеток. Показано, что один только HMGB1, без IL-1β, IL-18 и других DAMP, может вызывать воспаление.
Сигнальные липиды группы эйкозаиноидов, такие как лейкотриены и простагландины, увеличивают проницаемость кровеносных сосудов и способствуют воспалению. В 2012 было показано, что входящий ток кальция после активации каспазы 1 может индуцировать синтез эйкозаиноидов. При этом глицин, ингибирующий лизис клетки, но не секреторные пути, связанные с каспазой 1, не блокировал выделение эйкозаноидов, подтверждая, что они действительно секретируются, а не просачиваются во внешнюю среду пассивно при увеличении проницаемости мембраны. Сигнальные пути эйкозаиноидов не всегда связаны с активацией каспазы 1, однако они представляют собой очень быстрый и сильный провоспалительный сигнал, запускаемый каспазой 1.
В ходе ряда исследований было показано, что активация каспазы 1 и аутофагия антагонистичны по отношению друг к другу: активная каспаза 1 подавляет аутофагию, а аутофагия мешает активации каспазы 1 и уменьшает количество IL-1β и инфламмасом в цитозоле.
5.3. Пироптоз и патогены
Важность пироптоза как механизма, который удаляет патогенов из внеклеточной среды, для которой они приспособлены, подтверждается тем фактом, что у многих патогенов имеются механизмы, позволяющие им избежать этой реакции врождённого иммунитета. Среди таких патогенов есть как бактерии, так и вирусы. В общем, можно выделить три основных направления, на которых основаны эти механизмы избегания пироптоза.
- Патогены могут ограничивать экспрессию лиганда, активирующего инфламмасомы, компартментами, недоступными для инфламмасом, или делать так, чтобы лиганд не мог связаться с NLR.
- Патогены модифицируют структуру лиганда так, чтобы инфламмасомы не могли распознавать её;
- Патогены непосредственно блокируют функционирование инфламмасом.
Стоит подчеркнуть, что, в зависимости от патогена, для него может быть важным заблокировать не только пироптоз, но также секрецию интерлейкинов IL-1b и (или) IL-18 (или только её, без блокировки пироптоза).
В таблице ниже представлены стратегии, которые различные патогены используют для того, чтобы избежать пироптоза.
| Патоген | Стратегия | Эффектор | Инфламмасома/каспаза |
|---|---|---|---|
| Бактерии Salmonella typhimurium | Подавление экспрессии флагеллина (он активирует инфламмасомы) Модификация лигандов Поддержание стабильности вакуолей, в которых заключены бактерии | Флагеллин T3SS[en] SifA | NLRC4 NLRC4 NLRC4/NLRP3 |
| Francisella novicida[en] | Альтернативная структура LPS, не распознаваемая каспазой 11 Поддержание стабильности клеточной стенки | Тетраацетилированный LPS CRISPR-Cas | Каспаза 11 AIM2 |
| Listeria monocytogenes[en] | Подавление экспрессии флагеллина Ограничение экспрессии листериолизина О[en] (LLO) вакуолями | Флагеллин/MogR LLO | NLRC4 NLRP3 |
| Yersinia pseudotuberculosis[en] | Подавление активации каспазы 1 | YopM | Каспаза 1 |
| Legionella pneumophila | Поддержание стабильности вакуолей | ShdA | NLRC4, AIM2, каспаза 11 |
| Shigella flexneri[en] | Подавление активации каспазы 1 | OspC3 | Каспаза 4 |
| Вирусы Вирус миксоматоза[en] | Подавление активации каспазы 1 | M13L-PYD | Каспаза 1 |
| Вирус коровьей оспы[en] | Подавление активации каспазы 1 | CrmA | Каспаза 1 |
| Простой вирус герпеса саркомы Капоши[en] (KHSV) | Подавление инфламмасомы NLRP1 | Orf16 | NLRP1 |
Кроме вышеперечисленных бактерий, механизмами избегания пироптоза обладают такие бактерии, как Anaplasma phagocytophilum, Burkholderia thailandensis, Burkholderia pseudomallei. Кроме того, инфламмасомы связаны с развитием некоторых протозойных инфекций, например, вызванных Trypanosoma cruzi, Toxoplasma gondii, Plasmodium spp. и Leishmania.
6. Нетоз
Нето́з (англ. NETosis от англ. Neutrophil extracellular traps) — вид программируемой клеточной гибели, происходящей у нейтрофилов. Сопровождается выбрасыванием из погибающих нейтрофилов нитей, состоящих в основном из ДНК. Благодаря нетозу нейтрофилы убивают внеклеточных патогенов, минимизируя вред для других клеток.

Рис. (A-C) Анализ с помощью сканирующего электронного микроскопа срезов инфицированных Candida albicans легких мышей через 24 часа после интраназального заражения. (A) На изображении показана бронхиола, колонизированная C. albicans и инфильтрированная иммунными клетками хозяина, b = бронхиола. (B) Изображение с высоким разрешением области в рамке на (A) показывает респираторный эпителий бронхиол, колонизированных дрожжевой формой C. albicans (стрелка) и гифами (стрелка). (C) Изображение с высоким разрешением области в рамке на (B), показывающее внеклеточные ловушки нейтрофилов, покрывающие грибковые поверхности (стрелка). Шкала баров в (A) = 100 мкм, в (B) = 10 мкм и в (C) = 2 мкм.
6.1. Механизм
В условиях in vitro у нейтрофилов нетоз запускается под действием форболмиристатацетата, интерлейкина 8 (IL-8) и липополисахарида. При этом нейтрофилы высвобождают гранулы белков и хроматина (в основном эти гранулы состоят из ДНК), которые во внеклеточном пространстве формируют фибриллы (нити), опутывающие клетку патогена. Нетоз не всегда сопровождается гибелью нейтрофила: описан так называемый витальный нетоз, при котором ДНК упаковывается в везикулы, отшнуровывающиеся от клеточного ядра. Ниже будет описан механизм суицидального нетоза, при котором нейтрофил гибнет.
Молекулярный механизм запуска нетоза не до конца ясен, однако известны несколько ключевых белков этого процесса. Считается, что нетоз запускается при активации NADPH-оксидазы, которая начинает вырабатывать активные формы кислорода. Под их действием запускается фермент протеин-аргининдеиминаза 4 (PAD4). PAD4 вызывает цитруллинирование гистонов в ядре нейтрофила, в результате чего хроматин декомпактизируется. В ядро входят миелопероксидаза и эластаза нейтрофилов, которые стимулируют дальнейшую декомпактизацию хроматина и в конце концов приводят к разрушению ядерной оболочки. Деконденсированный хроматин выходит в цитоплазму, где к нему присоединяются дополнительные белки, формируя так называемые внеклеточные нити нейтрофилов (англ. Neutrophil extracellular traps, NETs). Образование NETs регулируется липоксигеназным путём. При некоторых способах запуска нетоза (например, при контакте с бактериальной клеткой) нейтрофил-5-липоксигеназа взаимодействует с фосфолипидами, и продукты реакции ингибируют образование NETs. Из внеклеточной среды NETs убирают макрофаги, которые фагоцитируют и разрушают их.
При суицидальном нетозе выбрасывание NETs сопровождается гибелью нейтрофилов посредством механизма, отличного от апоптоза и некроза. В случае суицидального нетоза после формирования внеклеточных нитей разрушается клеточная мембрана нейтрофила, в результате чего нити оказываются во внеклеточном пространстве. Суицидальный нетоз может быть запущен при активации Toll-подобных рецепторов (TLRs), Fc-рецепторов и рецепторов комплемента различными лигандами, такими как антитела или форболмиристатацетат. Считается, что при активации этих рецепторов в конечном счёте происходит выход ионов кальция из эндоплазматического ретикулума, который далее активирует NADPH-оксидазу. Процесс суицидального нетоза может занимать несколько часов, даже при воздействии высоких концентрации форболмиритатацетата, в то время как витальный нетоз происходит в течение нескольких минут.
Витальный нетоз запускается липополисахаридом и другими антигенами бактериального происхождения, TLR4-активированными тромбоцитами или белками комплемента совместно с лигандами TLR2. При витальном нетозе происходит блеббинг (отшнуровывание пузырьков) ядра, в результате чего в клетке появляются везикулы, заполненные ДНК, которую они выделяют наружу путём экзоцитоза без повреждения клеточной мембраны. Такие везикулы формируются и выбрасывают своё содержимое во внеклеточное пространство очень быстро, причём нейтрофил при этом не гибнет, хотя и остаётся без ДНК. Вопрос о том, можно ли считать живым нейтрофил без ДНК, является спорным. После витального нетоза нейтрофилы могут продолжить фагоцитировать и убивать клетки микроорганизмов.
