Клеточный цикл
| Site: | Электронный информационно- образовательный портал ВолгГМУ |
| Course: | Дисциплина Экспериментальная патобиохимия клетки |
| Book: | Клеточный цикл |
| Printed by: | Гость |
| Date: | Saturday, 18 May 2024, 4:20 PM |
Table of contents
1. Регуляция клеточного цикла
Закономерная последовательность смены периодов клеточного цикла осуществляется при взаимодействии таких белков, как циклин-зависимые киназы и циклины. Клетки, находящиеся в G0 фазе, могут вступать в клеточный цикл при действии на них факторов роста. Разные факторы роста, такие как тромбоцитарный, эпидермальный, фактор роста нервов, связываясь со своими рецепторами, запускают внутриклеточный сигнальный каскад, приводящий в итоге к транскрипции генов циклинов и циклин-зависимых киназ. Циклин-зависимые киназы становятся активными лишь при взаимодействии с соответствующими циклинами. Содержание различных циклинов в клетке меняется на протяжении всего клеточного цикла. Циклин является регуляторной компонентой комплекса циклин-циклин-зависимая киназа. Киназа же является каталитическим компонентом этого комплекса. Киназы не активны без циклинов. На разных стадиях клеточного цикла синтезируются разные циклины. Так, содержание циклина B в ооцитах лягушки достигает максимума к моменту митоза, когда запускается весь каскад реакций фосфорилирования, катализируемых комплексом циклин-В/циклин-зависимая киназа. К окончанию митоза циклин быстро разрушается протеиназами.
2. Фазы клеточного цикла
Кле́точный цикл — это период существования клетки от момента её образования путём деления материнской клетки до собственного деления или гибели

Клеточный цикл эукариот состоит из двух периодов:
- Период клеточного роста, называемый «интерфаза», во время которого идет синтез ДНК и белков и осуществляется подготовка к делению клетки.
- Период клеточного деления, называемый «фаза М» (от слова mitosis — митоз).
Интерфаза состоит из нескольких периодов:
- G1-фазы (от англ. gap — промежуток), или фазы начального роста, во время которой идет синтез мРНК, белков, других клеточных компонентов;
- S-фазы (от англ. synthesis — синтез), во время которой идет репликация ДНК клеточного ядра, также происходит удвоение центриолей (если они, конечно, есть).
- G2-фазы, во время которой идет подготовка к митозу.
- У дифференцировавшихся клеток, которые более не делятся, в клеточном цикле может отсутствовать G1 фаза. Такие клетки находятся в фазе покоя G0.
Период клеточного деления (фаза М) включает две стадии:
- кариокинез (деление клеточного ядра);
- цитокинез (деление цитоплазмы).
В свою очередь, митоз делится на пять стадий.
Описание клеточного деления базируется на данных световой микроскопии в сочетании с микрокиносъемкой и на результатах световой и электронной микроскопии фиксированных и окрашенных клеток.
2.1. Интерфаза G0
G0-фа́за, или фа́за поко́я, — период клеточного цикла, в течение которого клетки находятся в состоянии покоя и не делятся. G0-фаза рассматривается или как растянутая G1-фаза, когда клетка ни делится, ни готовится к делению, или как отдельная стадия покоя вне клеточного цикла[1]. Некоторые типы клеток, как, например, нервные клетки или клетки сердечной мышцы, вступают в состояние покоя при достижении зрелости (то есть когда закончена их дифференцировка), но выполняют свои главные функции на протяжении всей жизни организма. Многоядерные мышечные клетки, не подвергающиеся цитокинезу, также рассматриваются как пребывающие в G0-фазе. Иногда делается различие между клетками в G0-фазе и «покоящимися» клетками (в том числе нейронами и кардиомиоцитами), которые никогда не вступят G1-фазу, в то время как другие клетки в G0-фазе могут потом начать делиться.
Клетки вступают в G0-фазу с контрольной точки в G1-фазе, например, точки рестрикции у животных и стартовой точки у дрожжей. Это обычно происходит в ответ на нехватку факторов роста или питательных веществ. В течение G0-фазы аппарат клеточного цикла разобран, исчезают циклины и циклин-зависимые киназы. Клетка пребывает в G0-фазе до тех пор, пока не появится повод начать деление. Клетки некоторых типов в зрелом организме, как, например, паренхимные клетки печени и почек, вступают в G0-фазу почти навсегда, и побудить их вновь начать делиться могут лишь особые обстоятельства. Другие типы клеток, как, например, эпителиальные клетки, продолжают делиться в течение всей жизни организма и редко входят в G0-фазу.
Хотя многие клетки в G0-фазе могут умереть вместе со всем организмом, не все клетки, вступающие в G0-фазу, обречены в скором времени умереть. Это часто является результатом отсутствия стимула для клетки вновь вступить в клеточный цикл.

Старение клеток отличается от состояния покоя тем, что старение клеток — это состояние, вызванное повреждением ДНК или деградацией, делающей размножение клетки невозможным. Кроме того, старение, в отличие от покоя, часто служит биохимической альтернативой саморазрушению — апоптозу для сильно повреждённых клеток. Наконец, покой — обратимое состояние, а старение — нет
2.2. Митоз
На основании морфологических особенностей митоз условно подразделяется на стадии: профазу, прометафазу, метафазу, анафазу, телофазу. Первые описания фаз митоза и установление их последовательности были предприняты в 70—80-х годах XIX века. В конце 1870-х — начале 1880-х годов немецкий гистолог Вальтер Флемминг для обозначения процесса непрямого деления клетки ввёл термин «митоз»[3].
Продолжительность митоза в среднем составляет 1—2 часа[1][4]. Митоз клеток животных, как правило, длится 30—60 минут, а растений — 2—3 часа.[5] За 70 лет в теле человека суммарно осуществляется порядка 1014 клеточных делений[6].
Аппарат клеточного деления
Деление всех эукариотических клеток сопряжено с формированием специального аппарата клеточного деления. Активная роль в митотическом делении клеток зачастую отведена цитоскелетным структурам. Универсальным как для животных, так и для растительных клеток является двухполюсное митотическое веретено, состоящее из микротрубочек и связанных с ними белков. Веретено деления обеспечивает строго одинаковое распределение хромосом между полюсами деления, в области которых в телофазе образуются ядра дочерних клеток.
Ещё одна не менее важная структура цитоскелета отвечает за разделение цитоплазмы (цитокинез) и, как следствие, за распределение клеточных органелл. В животных клетках за цитокинез отвечает сократимое кольцо из актиновых и миозиновых филаментов. В большинстве клеток высших растений из-за наличия жёсткой клеточной стенки цитокинез протекает с образованием клеточной пластинки в плоскости между двумя дочерними клетками. При этом область образования новой клеточной перегородки определяется заранее предпрофазным пояском из актиновых микрофиламентов, а поскольку актин участвует также в формировании клеточных септ у грибов, возможно, что он направляет цитокинез у всех эукариот[19].
Веретено деления
Поздняя метафаза митоза в клетке лёгкого тритона (использованы иммунофлуоресцентные красители). Четко просматривается веретено деления, образованное микротрубочками (зелёные), и хромосомы (синие)
Формирование веретена деления начинается в профазе. В его образовании принимают участие полярные тельца (полюса) веретена и кинетохоры хромосом, и те и другие взаимодействуют с микротрубочками — биополимерами, состоящими из субъединиц тубулина. Главным центром организации микротрубочек (ЦОМТ) во многих эукариотических клетках является центросома — скопление аморфного фибриллярного материала, причём в большинстве животных клеток в состав центросом также входят пары центриолей[21]. Во время интерфазы ЦОМТ, как правило, располагающийся вблизи клеточного ядра, инициирует рост микротрубочек, расходящихся к периметру клетки и образующих цитоскелет. В S-фазе материал центросомы удваивается, а в профазе митоза начинается расхождение дочерних центросом. От них в свою очередь «отрастают» микротрубочки, которые удлиняются вплоть до соприкосновения друг с другом, после чего центросомы расходятся. Затем, в прометафазе, после разрушения ядерной мембраны, микротрубочки проникают в область клеточного ядра и взаимодействуют с хромосомами. Две дочерние центросомы теперь называют полюсами веретена[22].
По морфологии различают два типа митотического веретена: астральный (или конвергентный) и анастральный (дивергентный)[~ 1][24].
Астральный тип митотической фигуры, характерный для животных клеток, отличают благодаря небольшим зонам, на полюсах веретена, в которых сходятся (конвергируют) микротрубочки. Зачастую центросомы, располагающиеся в области полюсов астрального веретена, содержат центриоли. От полюсов деления также расходятся во всех направлениях радиальные микротрубочки, не входящие в состав веретена, а образующие звездчатые зоны — цитастеры.
Анастральный тип митотической фигуры отличается широкими полярными областями веретена, так называемыми полярными шапочками, в их состав не входят центриоли. Микротрубочки при этом расходятся широким фронтом (дивергируют) от всей зоны полярных шапочек. Этот тип митотической фигуры также отличает отсутствие цитастеров. Анастральный тип митотического веретена наиболее характерен для делящихся клеток высших растений, хотя иногда наблюдается и в некоторых клетках животных.
Микротрубочки
Микротрубочки — динамичные структуры, принимающие активное участие в построении веретена деления во время митоза. Химически они представляют собой биополимеры, состоящие из субъединиц белка тубулина. Количество микротрубочек в клетках различных организмов может значительно отличаться. В метафазе веретено деления в клетках высших животных и растений может содержать до нескольких тысяч микротрубочек, тогда как у некоторых грибов их всего около 40[22].
Митотические микротрубочки веретена деления «динамически нестабильны». Их «положительные» или «плюс-концы», расходящиеся во всех направлениях от центросом резко переходят от равномерного роста к стремительному укорочению, при котором часто деполимеризуется вся микротрубочка. Согласно этим данным образование митотического веретена объясняется селективной (выборочной) стабилизацией микротрубочек взаимодействующих в экваториальной области клетки с кинетохорами хромосом и с микротрубочками, идущими от противоположного полюса деления. Данная модель объясняет характерную двухполюсную фигуру митотического веретена[22].
Центромеры и кинетохоры
Основные статьи: Центромера, Кинетохор
Центромеры — специализированные последовательности ДНК, необходимые для связывания с микротрубочками веретена деления и для последующего расхождения хромосом. В зависимости от локализации различают несколько типов центромер. Для голоцентрических центромер характерно образование связей с микротрубочками веретена по всей длине хромосомы (некоторые насекомые, нематоды, некоторые растения). В противоположность голоцентрическим моноцентрические центромеры служат для связи с микротрубочками в единственной области хромосомы[24].
В центромерной области обычно располагаются кинетохоры хромосом — сложные белковые комплексы, морфологически очень сходные по своей структуре для различных групп эукариот, как, например, для диатомовых водорослей, так и для человека[25]. Обычно на каждую хроматиду (хромосому) приходится по одному кинетохору. На электронных микрофотографиях кинетохор обычно выглядит как пластинчатая трёхслойная структура[26]. Порядок слоев следующий: внутренний плотный слой, примыкающий к телу хромосомы; средний рыхлый слой; внешний плотный слой, от которого отходит множество фибрилл, образуя т.н. фиброзную корону кинетохора.
К основным функциям кинетохора относят: закрепление микротрубочек веретена деления, обеспечение движения хромосом во время митоза при участии микротрубочек, связывание между собой сестринских хроматид и регуляцию их последующего разделения в анафазе митоза[27]. Минимально достаточно одной микротрубочки (например, для дрожжей) ассоциированной с кинетохором, чтобы обеспечить движение хромосомы. Однако с одним кинетохором могут быть связаны целые пучки, состоящие из 20—40 микротрубочек (например, у высших растений или человека), чтобы обеспечить расхождение хромосом к полюсам клетки[26][27].
2.3. Циклины и циклин-зависимые киназы
Циклины — семейство белков-активаторов циклин-зависимых протеинкиназ (CDK) (англ. CDK, cyclin-dependent kinases) — ключевых ферментов, участвующих в регуляции клеточного цикла эукариот. Циклины получили своё название в связи с тем, что их внутриклеточная концентрация периодически изменяется по мере прохождения клеток через клеточный цикл, достигая максимума на его определенных стадиях.
Каталитическая субъединица циклин-зависимой протеинкиназы частично активируется в результате взаимодействия с молекулой циклина, которая образует регуляторную субъединицу фермента. Образование этого гетеродимера становится возможным после достижения циклином критической концентрации. В ответ на уменьшение концентрации циклина происходит инактивация фермента. Для полной активации циклин-зависимой протеинкиназы должно произойти специфическое фосфорилирование и дефосфорилирование определенных аминокислотных остатков в полипептидных цепях этого комплекса. Одним из ферментов, осуществляющих подобные реакции, является киназа CAK (CAK — CDK activating kinase).
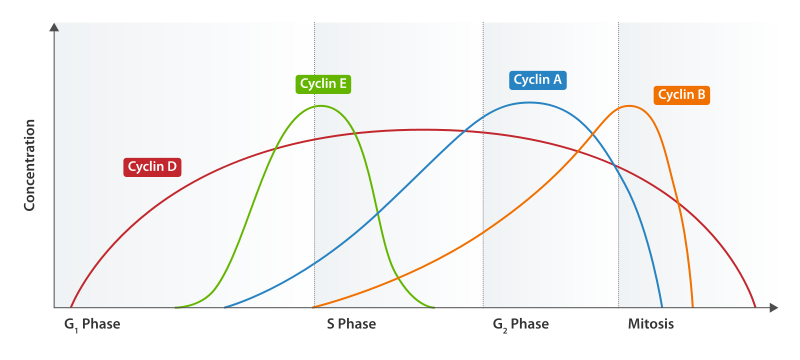
Рис. Концентрация циклинов в клеточном цикле
| циклин | Cdk | фаза клеточного цикла |
|---|---|---|
| циклин Е | Cdk2 | вход в S-фазу |
| циклин A | Cdk2 | вход в G2-фазу |
| циклин B | Cdk1 | вход в M-фазу |
|
Циклин зависимые киназы |
Субстраты |
|
|
CDKi (CDC2) |
A and B |
Lamins, histone Hi |
|
CDK2 |
E and A |
Rb, P107, P130, Cdt1, CP110 |
|
CDK3 |
C |
Rb |
|
CDK4 |
D |
Rb, P107, P130, SMAD2, and SMAD3 |
|
CDK6 |
D |
Rb, P107, P130, SMAD2, and SMAD3 |
|
CDK7 (CAK) |
H |
CDK1-CDK6, RNA pol 11 |
3. Контрольные точки клеточного цикла
Для определения завершения каждой фазы клеточного цикла необходимо наличие в нем контрольных точек. Если клетка «проходит» контрольную точку, то она продолжает «двигаться» по клеточному циклу. Если же какие-либо обстоятельства, например, повреждение ДНК, мешают клетке пройти через контрольную точку, которую можно сравнить со своего рода контрольным пунктом, то клетка останавливается и другой фазы клеточного цикла не наступает, по крайней мере, до тех пор, пока не будут устранены препятствия, не позволявшие клетке пройти через контрольный пункт. Существует как минимум четыре контрольных точки клеточного цикла: точка в G1, где проверяется интактность ДНК, перед вхождением в S-фазу, сверочная точка в S-фазе, в которой проверяется правильность репликации ДНК, сверочная точка в G2, в которой проверяются повреждения, пропущенные при прохождении предыдущих сверочных точек, либо полученные на последующих стадиях клеточного цикла. В G2-фазе детектируется полнота репликации ДНК, и клетки, в которых ДНК недореплицирована, не входят в митоз. В контрольной точке сборки веретена деления проверяется, все ли кинетохоры прикреплены к микротрубочкам.
5. Ингибиторы циклин-зависимой киназы
Ингиби́тор цикли́н-зави́симой кина́зы (англ. Cdk inhibitor protein, CKI, CDI, CDKI) — белок, блокирующий активность циклин-зависимой киназы отдельно или циклин-зависимой киназы в комплексе с циклином. Обычно сдерживающая активность CKI приурочена к фазе G1 клеточного цикла. К тому же, активация CKI может происходить в ответ на провреждения ДНК или может быть вызвана внеклеточными ингибирующими сигналами[2].
Большинство эукариотических организмов обладают ингибиторами циклин-зависимых киназ. В животных клетках выделяют два семейства CKI: Cip/Kip и INK4.

Ингибиторы семейства Cip/Kip блокируют циклин-зависимую киназу в комплексе с циклином, а ингибиторы семейства INK4 блокируют отдельные циклин-зависимые киназы Cdk4 и Cdk6. В животных клетках ингибиторы циклин-зависимых киназ разделяются на два основных семейства: Cip/Kip и INK4. Семейство Cip/Kip включает ингибиторы CDK белки p21, p27, p57. К основным субстратам Cip/Kip-ингибиторов относятся циклин-киназные комплексы G1/S-Cdk и S-Cdk, отвечающие, соответственно, за G1/S-переход и вступление в S-фазу. Ингибиторы семейства INK4 блокируют циклин-зависимые киназы Cdk4 и Cdk6 регулирующие G1-фазу клеточного цикла.
| Рис. Схема ингибирования Cdk6 с участием INK4. Белок INK4 соединяется с циклин-зависимой киназой Cdk6 и смещает аминоконцевую долю киназы примерно на 15° относительно оси вращения. В итоге, деформируется каталитическая область Cdk6, а также снижается способность циклин-зависимой киназы к связыванию циклина. |

{kind=link}
На протяжении фазы G1 в растущей клетке блокируется активность циклин-зависимых киназ (англ. Cdk) до момента вступления клетки в очередной клеточный цикл. Сдерживание активности Cdk обеспечивается тремя контрольными механизмами. Во-первых, снижением экспрессии генов циклинов. Во-вторых, увеличением степени деградации циклинов. Наконец, к третьему типу сдерживания активности Cdk относятся ингибиторы CKI. Помимо обеспечения стабильного роста клетки в фазе G1 ингибиторы циклин-зависимых киназ участвуют в аресте клеточного цикла на стадии G1 в ответ на неблагоприятные внешние условия. К тому же события клеточного цикла могут блокироваться с участием CKI при повреждениях ДНК[2].
Ингибиторы циклин-зависимых киназ: Sic1 у почкующихся дрожжей, Rum1 у делящихся дрожжей и Rux у Drosophila — несмотря на структурные различия обладают как минимум тремя сходными функциональными особенностями. Во-первых, основными мишенями данных CKI являются митотические циклин-киназы (англ. M-Cdk) и циклин-киназы синтетической фазы клеточного цикла (англ. S-Cdk). В то же время указанные ингибиторы CKI не могут блокировать циклин-зависимые киназы, обеспечивающие переход клетки из фазы G1 в S-фазу (англ. G1/S-Cdk). Наконец, третьей характерной особенностью всех перечисленных ингибиторов CKI является способ их деактивации. Все они разрушаются после фосфорилирования со стороны активных циклин-зависимых киназ[2].
Семейство Cip / Kip (p21, p27, p57) регулирует динамику актина посредством ингибирования пути Rho-ROCK-LIMK [2]

5.1. INK4 представляет собой семейство ингибиторов циклинзависимой киназы (CKI)
Члены этого семейства (p16INK4a, p15INK4b, p18INK4c, p19INK4d) являются ингибиторами CDK4 (отсюда их название - ингибиторы CDK4) и CDK6. Другое семейство CKI, белки CIP / KIP способны ингибировать все CDK. Усиленная экспрессия белков INK4 может привести к остановке G1, способствуя перераспределению белков Cip / Kip и блокируя активность циклина E-CDK2. Во время цикла в клетках происходит перераспределение белков Cip / Kip между CDK4 / 5 и CDK2 по мере того, как клетки проходят через G1. [1] Их функция, ингибирующая CDK4 / 6, состоит в том, чтобы блокировать прогрессирование клеточного цикла за пределами точки ограничения G1. [2] Кроме того, белки INK4 играют роль в клеточном старении, апоптозе и репарации ДНК. [3]
Белки INK4 являются опухолевыми супрессорами, а мутации потери функции приводят к канцерогенезу. [4]
Белки INK4 очень похожи по структуре и функциям, с аминокислотным сходством до 85%. [1] Они содержат несколько анкириновых повторов. [3].
Каждый повтор состоит из расширенной цепи, соединенной мотивом спиральная петля-спираль (HLH) со следующей расширенной цепью. Кристаллическая структура комплекса p19Ink4d-CDK6 была решена и предоставила ценные сведения о механизме ингибирования CDK белками чернил (рис. 11-4) .17 α-спирали и β-витки анкириновых повторов p19Ink4d образуют «шапку» над N-концевой домен CDK6 и вызывают его пространственное перемещение от С-конца. Это событие ингибирует продуктивное связывание АТФ, но не мешает образованию комплекса CDK-циклин. Как и ожидалось из их структуры, все четыре белка Ink проявляют сходную биохимическую активность по отношению к CDK4 и CDK6. Интересно, что короткий пептид, который был получен из одного из мотивов анкирина, обладал способностью связывать и ингибировать CDK4, подразумевая важность этих доменов в функциональности Ink4.
Несмотря на сходную биохимическую активность и сопоставимые третичные структуры белков Ink, их регуляция различна. P16Ink4a не экспрессируется в большинстве тканей. Скорее, он индуцируется в ответ на экспрессию онкогенных или трансформирующих белков и во время клеточного старения. Несколько онкогенов, а также опухолевых супрессоров регулируют экспрессию p16Ink4a. Например, избыточная экспрессия Ras увеличивает уровни p16Ink4a в первичных клетках грызунов. 19 Инактивация белка, чувствительного к ретинобластоме, Rb или опухолевого супрессора p53 также может стимулировать экспрессию p16Ink4a. 20 Напротив, экспрессия p15Ink4b регулируется факторами, ингибирующими рост ( Митогены), такие как TGF-β. По-видимому, только экспрессия p18Ink4c и p19Ink4d регулируется на различных фазах клеточного цикла, причем пик экспрессии наблюдается на S-фазе.21 Характер экспрессии белков Ink4 также дифференцированно регулируется во время развития.
Kim WY, Sharpless NE (October 2006). "The regulation of INK4/ARF in cancer and aging". Cell. 127 (2): 265–75. doi:10.1016/j.cell.2006.10.003. PMID 17055429.
Ortega S, Malumbres M, Barbacid M (March 2002). "Cyclin D-dependent kinases, INK4 inhibitors and cancer". Biochimica et Biophysica Acta. 1602 (1): 73–87. doi:10.1016/S0304-419X(02)00037-9. PMID 11960696.
Cánepa ET, Scassa ME, Ceruti JM, Marazita MC, Carcagno AL, Sirkin PF, Ogara MF (July 2007). "INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions". IUBMB Life. 59 (7): 419–26. doi:10.1080/15216540701488358. PMID 17654117.
Roussel MF (September 1999). "The INK4 family of cell cycle inhibitors in cancer". Oncogene. 18 (38): 5311–7. doi:10.1038/sj.onc.1202998. PMID 10498883
5.2. CDKN1A или P21
CDKN1A (англ. cyclin-dependent kinase inhibitor 1A, p21, Cip1) — внутриклеточный белок-ингибитор циклин-зависимой киназы 1A, играет критическую роль в клеточном ответе на повреждение ДНК. Уровень белка повышен в клетках, находящихся в стадии покоя, таких как дифференцированные клетки организма. Один из 9 известных белковых ингибиторов циклин-зависимой киназы.
p21 обеспечивает устойчивость гематопоэтических клеток к инфицированию ВИЧ за счёт связывания с вирусной интегразой, предотвращая таким образом встраивание провируса в хромосомный аппарат клетки.

Ген CDKN1A как правило не инактивируется полностью в злокачественных опухолях. Точная роль p21 в канцерогенезе до конца пока не установлена. Исследования показывают, что при некоторых типах опухолей потеря p21 является признаком плохих шансов на выживание. Однако известны ситуации, когда повышенная концентрация этого белка в клетках положительно коррелирует с агрессивностью опухоли и её способностью к метастазированию. Это особенно относится к тем случаям, когда p21 накапливается в цитоплазме, а не в ядре клетки
В природе регенеративные способности (восстановление тканей, органов и конечностей) наиболее выражены у амфибий, полипов, иглокожих, губок, гидр, дождевых червей, саламандр и нек. др., в то время как у млекопитающих они существенно ограничены.

В лаборатории молекулярного биолога Хэбер-Катц регенерацией у млекопитающих заинтересовались в 1996 году, когда во время изучения одного из аутоиммунных заболеваний зафиксировали у лабораторных мышей линииMurphy Roths Large (MRL) восстановление хрящевой ткани ушей после пожизненной маркировки (перфорации). У животных других линий такой регенерации не происходило. Ученые исследовали восстановленные участки и зафиксировали в них синтез ДНК, пролиферацию клеток и появление новых волосяных фолликулов.
На фото MRL линия мышей справа. Контрольная группа мышей слева. http://news.bbc.co.uk/hi/spanish/science/n
Последующие эксперименты на мышах MRL показали, что после нанесенных им повреждений в области сердечной мышцы и спинного мозга, эти ткани так же успешно регенеририруют без образования рубцов.
Чтобы с наибольшей вероятностью вычислить гены, ответственные за процессы регенерации у мышей MRL, было решено картировать их геном. Так же под прицел ученых попал феномен влияния изменений в ДНК на цикл деления клеток.
Исследования показали, что у животных MRL линии неактивен ген р21, играющий важную роль в регуляции клеточного деления и в торможении процесса деления клеток при повреждении ДНК.
В новой работе ученые показали, что мыши без гена p21 демонстрируют регенеративные характеристики, наблюдаемые у мышей MRL линии. У генноинженерных мышей фиксировалось быстрое деление клеток, восстанавливающее ткани и быстрый апоптоз - механизм клеточного самоуничтожения, включающийся, если ДНК повреждена. Сочетание этих процессов свойственно видам, обладающим регенеративными способностями. Ученые считают, что такое совокупное воздействие позволяет клеткам данных видов делиться быстро, не выходя из-под антиракового контроля.
Профессор Хебер-Катц о захватывающих перспективах исследования (газета Guardian): "Как тритон, потерявший конечность, эти мыши могут заменить отсутствующие или поврежденные ткани здоровыми, без единого следа шрамов. Мы только начинаем осознавать последствия этих открытий, но возможно, когда-нибудь мы сможем ускорять выздоровление человека, временно отключая ген p21».
"Мы полагаем, что отключение гена p21 приводит к быстрой регенерации у млекопитающих. Подобная структура ДНК присутствует у всех видов животных, способных восстанавливать части тела. Так что если нам удастся проверить нашу теорию на людях и разработать способ временного "отключения" этого гена, вполне вероятно, что медики смогут "выращивать" утраченные конечности пациентов".
Эллен Хэбер-Катц – профессор молекулярной и клеточной онкологии Университета Вистара (США)
heberkatz@wistar.org.
Страница: www.wistar.org/research_facilities/heber

Ссылки: статья Lack of p21 expression links cell cycle control and appendage regeneration in mice/ Proceedings of the National Academy of Sciences.
В научной публикации, опубликованной в Трудах Национальной академии наук, коллектив Эллен Хэбер-Катц предоставляет веские доказательства, что тканевая регенерация связана с контролем клеточного деления.
5.3. Заключение
| Фазы |
|  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Регуляторы |
|
6. Сигнальные пути, регулирующие клеточный цикл
По крайней мере 3 пути
- ATM-Chk2 и ATR-Chk1 пути
- p21-активируемая киназа 1 (PAK1) пути
- MAP киназа сигнальные пути
6.1. ATM-Chk2 и ATR-Chk1 пути
Одним путем, регулирующим прогресс клеточного цикла является путь АТМ-Chk2, который может остановить клеточный цикл на многочисленных контрольных точках, если повреждение ДНК обнаружено. В нормальных клетках, когда в двухцепочечной имеется разрыв обнаружено, что основным активатором ответного пути является атаксия-телеангиэктазия-мутантная (ATM), протеинкиназа. АТМ фосфорилирует следующие белки каскада, обладающие консенсусными последовательностями, содержащие остатки либо серина или треонина, непосредственно предшествующие остатку глутамина. Nbs1 усиливает активность ATM для усиления ответа. Одним из белков, фосфорилированных в остатке треонина в положении 68 с помощью ATM является киназа Chk2, которая распространяется сигнал также путем фосфорилирования белков вниз по каскаду. Мишенью Chk2 является cdc25, который после фосфорилирования становится либо мишенью для деградации или секвестрации в цитоплазме, тем самым вызывая остановку клеточного цикла в G1, S и G2 / M стадии. Chk2 участвует в регуляции транскрипции, а также, с помощью фосфорилирования E2F-1 и р53, способствуя транскрипции генов в каскадах их ответов. Аналогичный сигнальный путь, который реагирует на присутствие одноцепочечной ДНК является ATR-Chk1 путь. ATR-Chk1 активируется если репликация ДНК затруднена, например, когда существует дефицит нуклеотидов или УФ-индуцированные димеры, препятствующие генерации репликативной вилки, что приводит к образованию ssДНК. Этот путь обычно активируется в сочетании с каскадом АТМ-Chk2 (16). TopBP1 усиливает активность ATR неизвестным механизмом, который затем фосфорилирует Claspin, который в свою очередь связывается с Chk1. Chk1 затем ассоциируется в ATR, поэтому он может быть фосфорилирован по остаткам серина, и, следовательно, ингибирует Сdc25 путем фосфорилирования и тем самым способствует его деградации. Таким образом, этот путь вызывает торможение клеточного цикла. Эти два пути могут замедлить прогрессирование от G1 до фазы S, действовать в рамках фазы S, чтобы замедлить процесс репликации ДНК, или предотвратить прогрессирование от G2 до митоза. Таким образом, оба пути могут действовать в любой момент до митоза, чтобы предотвратить разделение, если есть повреждение ДНК. Существуют также контрольные точки в рамках процесса репликации ДНК, чьи ответы можно управлять обоими путями. Эти ответы включают стабилизирующие застопорился вилки репликации, подавление репликации в начале координат, а также задержки вступления в митоз, пока репликация ДНК не будет полностью завершена.
Поврежденная система реагирования на повреждения ДНК с участием как ATM-Chk2 и пути ATR-Chk1 часто встречается при многих видов рака. Это приводит к неспособности остановить клеточный цикл при необходимости, то есть утрате возможности регулирования клеточного цикла на контрольно точках, а также клетки, несущие двойные разрывы (DSBs) и другие аномалии ДНК способны прогрессировать через цикл и делиться, распространяя вниз мутации. Такие мутации предрасполагает индивидуумов к лимфоме (Smith J, Tho LM, Xu N, Gillespie DA. 2010. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Advances in Cancer Research. 108:73-112). ATM считается частично пенетрантный ген, обусловливающий предрасположенность к раку, известно, что мутации в этом гене повышает частоту рака, связанного с воздействием мутагенов окружающей среды (Ibid). Мутации в Nbs1 также связаны с предрасположенностью к раку. Связь Chk2 полиморфизма с раком груди и простаты определяется в диапазоне от умеренной до слабой (Ibid). С другой стороны, была выдвинута гипотеза, что Chk1 необходима для пролиферации специфических раковых стволовых клеток, таких, как эпидермальные клетки, порождающие опухоли кожи, вызванных воздействием канцерогенов. Интересно отметить, что удаление Chk1 в эпидермальных клетках мыши на самом деле подавляет канцероген-индуцированных туморогенез кожи. Клетка или организм может выдержать потерю сигнализации ATM-Chk2, при этом они могут быть предрасположены к развитию рака (ibid). Путь АТР-Chk1 необходим для клеточной пролиферации и выживании во многих типах клеток. Таким образом, в качестве терапевтических стратегий борьбы с туморогенезом может выбрать воздействие либо на ATR или на Chk1. Тем не менее, мышиные модели также показали, что частичная потеря функции Chk1 может способствовать туморогенезу, но необходимы дальнейшие исследования в этой области.
Терапевтический препарат тамоксифен способен блокировать Chk1 в эпидермальных клетках, снижая превращение папиллом кожи к карциному (Olshavsky N.A., Groh E.M., Comstock C.E., Morey L.M., Wang Y., Revelo M.P., Burd C., Meller J., Knudsen K.E., (2008) Cyclin D3 action in androgen receptor regulation and prostate cancer. Oncogene, 27(22):3111-21). Большинство терапевтических стратегий были сосредоточены на воздействии на Chk1, так как он является сильным эффектором повреждения ДНК и активен в большинстве опухолей. Chk1 ингибирование увеличивает повреждение в клетках за счет увеличения DSBs, тем самым активируя пути апоптоза. Если нормальные клетки преждевременно вступают в митоз (то есть в них увеличивается содержание нереплицированных ДНК) из-за невыполнения требований различных контрольных точек клеточного цикла, они должны погибнуть. Ингибирование Chk1, ATM и ATR также усиливает эффекты генотоксических агентов, включая излучения и аналоги нуклеозидов. XL-844 представляет собой новый разрабатываемый ингибитор Chk2, который будет блокировать контрольные точки S фазы, что приводит к преждевременному входу в митоз, и следовательно снижению выживаемости.
http://www.cubocube.com/dashboard.php?a=1642&b=1691&c=1
 |
|---|
| Chk1 и Chk2 киназы являются серин/треонин киназами, которые активируются киназами ATM и ATR в ответ на повреждения ДНК. Пусковым моментом для киназ являются преобразователи сигнала повреждения ДНК. и оба фермента фосфорилируют ряд субстратов, участвующих в реакции на повреждения ДНК. Chk1 и Chk2 имеют ряд перекрывающихся субстратов, хотя очевидно, что они имеют различные роли в управлении ответом клетки на повреждение ДНК. По современным представлениям киназы контрольной точки участвуют не только в регуляции клеточного цикла, но и в других аспектах клеточного ответа на повреждение ДНК. G1 контрольная точка модулируется в основном по ATM-Chk2-p53 пути, в то время как экспрессия ATR, Chk1 и CDC25A ограничена, пока клетка не проходит эту точку рестрикции. В другой момент, уровни ATR, Chk1 и CDC25A возрастают. Если повреждение ДНК обнаружено, Chk1/Chk2 активируются, CDC25A фосфорилируется, и, таким образом, дестабилизируется, в результате чего наблюдается p53-независимое торможение S периода. В фазе S, тот же самый каскад может привести к внутренней остановке клеточного цикла в S стадии в ответ на торможение образования вилок репликации. G2-M контрольной точки предотвращает попадание в митоз неустраненных повреждений ДНК. Начало этой контрольной точки опосредуется в каскадах ATM/ATR/Chk1/Chk2, как это показано, что в конечном итоге подавляет промитотическую активность циклин B/cdc2. Наряду с их центральной роли в модуляции контрольных точек клеточного цикла, Chk1 и Chk2 также участвуют в других аспектах реакции повреждения ДНК, включая репарации ДНК, индукции апоптоза и хроматина. |

Molecular organization of the checkpoint signaling network in response to DNA DSBs and replicative stress. Functional components of the checkpoint signaling network are hierarchically categorized as sensors, proximal transducer kinases, mediators, distal transducer kinases, and effectors. ATM is believed to be the primary proximal transducer kinase reactive to DNA DSBs. ATM activation via the formation of autophosphorylated monomers may be caused by changes in higher chromatin structure associated with DSBs. Current data suggest that recruitment of activated ATM to the site of damage requires the Mre11-Rad50-NBS1 complex. Phosphorylation of ATM substrates is facilitated by mediator molecules, such as 53BP1, Brca1, and MDC1, which themselves are often modified by ATM. In response to replicative stress, the ATR–ATR-interacting protein (ATRIP) complex is recruited to the region of ssDNA coated by recombinant protein A (RPA). Activation of Chk1 by ATR also requires the mediators Claspin and TopBP1 and loading of the sliding clamp Rad9-Rad1-Hus1 (“9-1-1”) complex onto DNA by the RFC complex. Signals originating from the sites of damage are amplified through the mobile distal transducer kinases Chk1/Chk2, which can disperse the damage alert to the rest of the cell. Both Chk1 and Chk2 can phosphorylate a variety of effectors (e.g., cdc25 phosphatases and p53), which ultimately halt cell cycle progression by inhibiting the cell cycle engines [cyclin-dependent kinases (cdk)]. Phosphorylation of cdc25A and cdc25C by Chk1/Chk2 results in enhanced degradation and cytoplasmic sequestration of the phosphatase, respectively. In addition to causing cell cycle delay, Chk1/Chk2 also mediates other aspects of the DNA damage response, including DNA repair, apoptosis, and chromatin remodeling by phosphorylating various downstream substrates (see text). A newly reported route of signaling (broken line) involves an ATM-dependent activation of the ATR-Chk1 pathway in response specifically to DSBs induced by IR. Key activating and inhibitory phosphorylation sites (green and red, respectively).
6.2. PAK1 путь
Еще одним сигнальным путем, который изменяется в раковых клетках, является p21-активированная киназа (PAK). Этот сигнальный путь связан с регулированием таких клеточных процессов как включая активность факторов роста и стероидных рецепторов, реорганизации цитоскелета, опухолевой трансформации и выживаемости клеток (Eswaran J, Li DQ, Shah A, Kumar R. 2012. Molecular pathways: targeting p21-activated kinase 1 signaling in cancer: opportunities, challenges, and limitations. Clinical Cancer Research. 18:3743-3749).
 |
|---|
| Схематическое изображение, показывающее несколько сигнальных путей, приводящих к активации РАК1 через G-белки и тирозин-киназы-зависимых и -независимые механизмы. Наиболее хорошо изученным путь для активации PAK1 является при участии Rho GTPases, RAC1 и цикла клеточного деления 42 (CDC42). Рецепторные тирозинкиназы (через рецептор фактора роста-связанного белка 2 (Grb2) и NCK), тирозинкиназы без рецепторов, такие как ЕТК, липиды, интегринами и вверх по течению серин/треонин киназ, таких как фосфатидилинозитол 3-киназы (PI3K) и пируват-дегидрогеназы киназы изофермента 1 (PDK1) также может активировать PAK1. Активация PAK1 также происходит Повсеместно взаимодействия партнеров, которые выпускают автоматическое торможение путем связывания с PAK1. Активированный PAK1 в свою очередь, инициирует каскад путей, которые достигают высшей точки в клеточном ответе. Как следствие выше функциональной активности Paks в опухолях, либо в силу повышенной экспрессии или активности, клетки приобретают определенное преимущество и показать характеристики, которые связаны с малигнизацией |
PAK1 активируется внешними стимулами, обнаруженных активирующих белков в плазматической мембране, ведущей к каскаду, что приводит к фосфорилированию РАК1, который переходит из неактивного состояния в активное конформации. В основе активации PAK1 в раковых клетках лежит не мутация. Она действует как онкоген. Это может произойти в результате увеличения числа копий гена, ингибирование его негативных регуляторов (p53) или гиперактивации ГТФаз включая Rac и Cdc42.

PAK1 увеличивает тирозинкиназную активность после того, как на плазматической мембране формируются комплексы молекул с помощью адаптерного белка NCK при действии факторов роста. Таким образом, он является важным компонентом для онкогенеза, который регулируется передачей сигналов от факторов роста, и он участвует в процессах клеточного роста, пролиферации и дифференцировки, Срыв PAK1 опосредованной передаче сигналов фактора роста является общей характеристикой раковых клеток.
Цитоскелет также является важным участником клеточного цикла. PAK1 специфически влияет на клеточный цикл через фазу M. Исследование, выполненное в раковых клетках молочной железы показали, что сверхэкспресия PAK1 привела к формированию многочисленных полюсов веретена и к неправильному разделению хромосом, событие, известный как анеуплоидии, который показан на фиг

Хотя PAK1 участвует в нескольких длинных сигнальных каскадах для других клеточных процессов, сигнальный путь регулирующий клеточный цикл довольно короткий. PAK1 обычно локализуется на полюсах веретена в ядре в результате своей киназной активности , где он может или фосфорилировать резидентные полюсные молекулы Aurora A или Arpc1b по остаткам треонина в положении 21. Arpc1b также может быть фосфорилирован Aurora A, после того, как последний фосфорилируется РАК1. Aurora A киназа участвует в делении клеток и их генетического материала путем контроля сегрегации хроматид. Он функционирует во время профазы митоза и необходим для правильного функционирования центросом ( Keen N, Taylor S. 2004. Aurora-kinase inhibitors as anti-cancer agents. Nature Reviews Cancer. 4:927-936). Дефекты в его функции вызывают генетическую нестабильность и приводят к онкогенезу. Кроме того, PAK1 фосфорилирует кофактора B тубулина, белок, ответственный за дестабилизацию микротрубочек, которая важна для изменений цитоскелета, происходящих во время деления. Вовлечение и локализация РАК1 в ядре имеет важное значение для успешного завершения митоза, а также в пролиферации раковых клеток.
Значительные усилия были предприняты для разработки терапевтических агентов против раковых клеток сверхэкспрессирующих PAK1. Эндогенный терапевтический подход должен был бы увеличить экспрессию гена NF2 супрессорf опухолей, который ингибирует активацию РАК1 путем связывания с его p21-связывающим доменом. МикроРНК-7 может также ингибировать PAK1 путем связывания с его 3 'нетранслируемой области. CEP-1347 представляет собой низкомолекулярный ингибитор, который блокирует функцию PAK1 через прямое связывание. И, наконец, можно также использовать аллостерические ингибиторы, такие как IPA-3, так как они ковалентно связывают PAK1 регулирующий домен, блокируя его способность связывать и активировать Cdc42. Другие подходы, включая использование металлоорганических соединений проходят испытания.
6.3. Map киназный сигнальный каскад
Сигнальные пути MAPK (англ. mitogen-activated protein kinase — митоген-активируемая протеинкиназа) — группа мультифункциональных внутриклеточных сигнальных путей, содержащих одну из митоген-активируемых протеинкиназ и контролирующих транскрипцию генов, метаболизм, пролиферацию и подвижность клеток, апоптоз и другие процессы
Сигнальные пути MAPK консервативны у эукариот и содержат характерный модуль, состоящий из трёх протеинкиназ. Эти пути активируются внеклеточными сигналами, такими как гормоны, факторы роста, хемокины и нейротрансмиттеры, которые распознаются соответствующими рецепторными тирозинкиназами или рецепторами, ассоциированными с G-белками. Рецепторы активируют ГТФазы семейств Ras и Rho. ГТФазы передают сигнал на модуль, состоящий из киназы киназы митоген-активируемой киназы (англ. MAPK kinase kinase, MKKK), которая фосфорилирует и активирует киназу митоген-активируемой киназы (англ. MAPK kinase, MKK), которая, в свою очередь, активирует митоген-активируемую киназу. MAPK фосфорилируют белки-мишени по остаткам серина и треонина и таким образом передают сигнал дальше. Кроме киназ, в состав сигнальных путей входят протеинфосфатазы и белки, которые обеспечивают сборку белковых комплексов
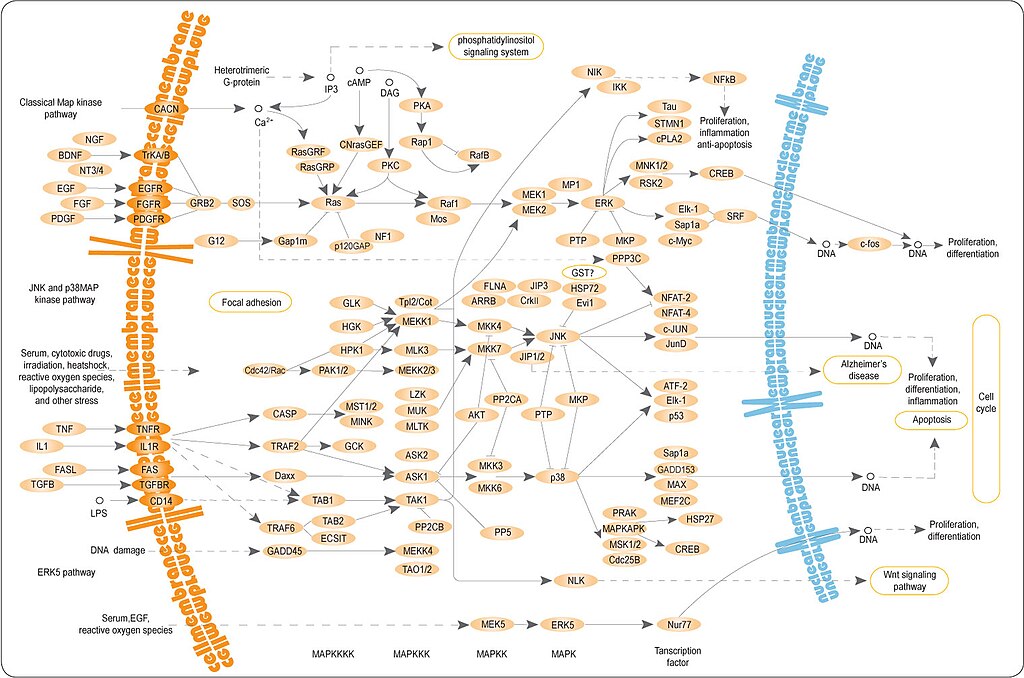
Классификация
У млекопитающих известно 4 основных MAPK-сигнальных пути:
- пути ERK (англ. extracellular signal-regulated kinase),
- ERK5 (англ. extracellular signal-regulated kinase 5),
- JNK (англ. c-Jun N-terminal kinase) и
- p38.
Как правило, сигнальные пути ERK отвечают на факторы роста, в то время как JNK и p38 реагируют на внеклеточные стрессовые сигналы. Такие же пути были обнаружены у Drosophila и Caenorhabditis elegans. Однако у млекопитающих эти пути более сложно устроены за счёт того, что MAP-киназы представлены не одним ферментом, а группой близких по структуре ферментов, которые кодируются несколькими генами (например, ERK1, ERK2 и др.). Кроме того, дополнительное разнообразие ферментов порождается альтернативным сплайсингом.

Сигнальный путь ERK (Ras-ERK, MAPK/ERK) — это один из ключевых и наиболее хорошо изученных сигнальных путей MAPK (англ. mitogen-activated protein kinase). Своё название этот путь получил по центральной MAP-киназе ERK (англ. extracellular signal-regulated kinase), которая представлена двумя близкими по структуре белками, ERK1 и ERK2. Данный путь может быть активирован внеклеточными сигналами, такими как гормоны, факторы роста, хемокины и нейротрансмиттеры, которые распознаются соответствующими рецепторными тирозинкиназами или рецепторами, ассоциированными с G-белками. Передача сигнала по ERK-пути в конечном итоге приводит к выживанию, пролиферации и увеличению подвижности клеток.
Как было сказано выше, сигнальный путь ERK может быть активирован в ответ на сигналы, полученные клеткой через рецепторные тирозинкиназы или рецепторы, сопряжённые с G-белками. Около цитоплазматической части таких рецепторов собирается сигнальный комплекс из множества белков, который в конце концов активирует ГТФазу Ras. Ras связывает и активирует киназу киназы MAPK/ERK (MAPK/ERK kinase kinase или MEKK), главными компонентами которой являются белки семейства Raf (Raf-1, A-Raf и B-Raf). MEKK фосфорилирует и активирует киназу MAPK/ERK (MAPK/ERK kinase или MEK), представленную двумя компонентами MEK1 и MEK2. MEK1/2 активирует ERK1/2.

Фосфорилирование ERK1/2 происходит вблизи клеточной мембраны[1]. После этого фермент диффундирует в цитоплазму, где фосфорилирует сигнальные белки, в том числе p90 киназу рибосомального белка S6 (p90 ribosomal S6 kinase или RSK), а затем в ядро, где он регулирует транскрипцию. ERK1/2 индуцирует транскрипцию ранних генов c-Fos и c-Myc, продукты которых являются факторами транскрипции и обеспечивают транскрипцию поздних генов, ответственных за пролиферацию, выживание и подвижность клеток[2].
Сигнальный путь ERK принимает участие в активации T-клеток, пролиферации эндотелиальных клеток при ангиогенезе, в регуляции синаптической пластичности и фосфорилировании транскрипционного фактора p53
Роль Raf-MEK-ERK наиболее хорошо изучена в патогенезе опухолей

После стимуляции рецептора EGF факторами роста, он димеризуется, что позволяет ему связать Grb2 на своих внутриклеточных доменах. Grb2 затем ассоциируется Sos, который способен активировать Ras, который затем инициирует каскад фосфорилирования МАР-киназ приводит к фосфорилированию и активации ERK (MAPK). Фосфорилированные ERKs могут образовывать гомодимеры и транслокации в ядро, где они активируют различные факторы транскрипции, приводящих к изменению экспрессии генов. Часто путь Raf-MEK-ERK приводит к усилению клеточной пролиферации (19, 21). Как видно на рисунке, как Ras и Raf часто оказываются мутировать в результате чего их конститутивной активации в различных видах опухолей (21). Ras белки функционируют как ВВП / GTP-регулируемых выключателей с внутренней GTPase активностью. Обмен ВВП к GTP приводит к активации Ras обусловлено факторами обмена гуанин нуклеотидных (RasGEFs е. Г. СЦ), в то время как гидролиз GTP к GDP, вызывающего инактивацию производится самой Ras через свою активность ГТФ. Мутировали Ras белки часто теряют способность гидролизовать ГТФ, оставляя РАС в стимула-независимого активированном состоянии, в то время как Raf, как известно, является сильным ретровирус онкоген (21). Несколько ингибиторов Ras, Raf и MEK были разработаны и показали большие перспективы. Благодаря высокому возникновению лекарственной устойчивости раковых клеток, два ингибитора (д. Ж. Один против Raf и один против МЕК) часто используются в комбинации (21). На сегодняшний день ни одного успешного ингибитора ERK разработано не было (21)