Книга_Физические методы
| Сайт: | Электронный информационно- образовательный портал ВолгГМУ |
| Курс: | Дисциплина Медицинская биохимия, принципы измерительных технологий в биохимии, патохимия, диагностика, биохимия злокачественного роста Часть 1 |
| Книга: | Книга_Физические методы |
| Напечатано:: | Гость |
| Дата: | Среда, 3 Июль 2024, 17:19 |
Оглавление
1. Гравиметрический анализ
Гравиметрический анализ (гравиметрия, весовой анализ) — метод количественного химического анализа, основанный на точном измерении массы вещества. Использует закон сохранения массы веществ при химических превращениях. Сыграл большую роль в становлении закона постоянства состава химических соединений, закона кратных отношений, периодического закона и др. Применяется для определения химического состава различных объектов (горных пород и минералов), качества сырья и готовой продукции, содержания кристаллизационной воды в солях, зольности топлива и так далее.
К достоинствам гравиметрического анализа относят высокую точность (обычно погрешность составляет 0,1—0,2 %) и отсутствие необходимости в предварительной градуировке измерительных приборов. С другой стороны, его проведение зачастую более трудоёмко и занимает больше времени по сравнению с другими методами
1.1. Методы анализа
Методы осаждения
Методы осаждения — наиболее распространённые методы гравиметрического анализа[2][3]. Навеску анализируемого вещества растворяют в воде или другом растворителе и осаждают определяемый элемент реактивом в виде малорастворимого соединения. Полученный осадок отфильтровывают, промывают, высушивают, прокаливают и взвешивают. По массе осадка после прокаливания вычисляют массовую долю определяемого элемента в навеске.
Поскольку осаждённое вещество может не соответствовать тому, что получается после прокаливания, различают осаждаемую и гравиметрическую (весовую) форму осадка. Осаждаемая форма — это соединение, осаждающееся из раствора при взаимодействии с соответствующим реагентом в процессе проведения анализа[4], а гравиметрическая форма образуется из осаждаемой при высушивании или прокаливании[5].
Например:

![{\displaystyle {\ce {CaC2O4 ->[t] CaO + CO2 ^ + CO ^}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f90d1ef258cb02969a7da4d54563a5195470fa7a)
В данном случае CaC2O4 является осаждаемой формой, CaO — гравиметрической формой.
В качестве осадителей применяют как неорганические, так и органические реагенты. Например, серу в виде сульфат-ионов осаждают ионами бария[6], железо осаждают раствором аммиака[7], а для осаждения алюминия часто используют 8-оксихинолин[8].
Методы отгонки[править | править код]
В этих методах определяемый компонент выделяют в виде летучего соединения действием кислоты или высокой температуры. Методы отгонки делятся на прямые и косвенные:
- прямые методы: определяемый компонент выделяют в виде летучего соединения и поглощают поглотителем (либо конденсируют). Расчёт ведут по изменению массы поглотителя.
- косвенные методы: вещество взвешивают, отгоняют летучее соединение и вновь взвешивают. Расчёт производят по уменьшению массы навески[2].
Методы выделения[править | править код]
Также существуют методы, основанные на выделении определяемого компонента из анализируемого вещества и его точном взвешивании, например восстановление ионов меди до металла с последующим взвешиванием[9].
1.2. Подготовка пробы, взятие и растворение навески
Сначала берётся средняя проба, состав которой отражал бы состав исследуемого материала. Средняя проба должна быть составлена из как можно большего числа проб, взятых из случайных точек материала. Для дальнейшей подготовки используется метод квартования: пробы тщательно измельчают, перемешивают, затем выкладывают в форме квадрата, делят диагоналями, после чего 2 противоположных треугольника отбрасывают, а другие снова смешивают, и так до тех пор, пока не останется порядка 25 г вещества[10].
После получения средней пробы берётся навеска. Как правило, чем больше навеска, тем выше точность определения, однако получающийся большой осадок трудно профильтровать, промыть и прокалить. Опытным путём установлено, что наиболее удобны в работе кристаллические осадки массой около 0,5 г и объёмистые аморфные осадки массой 0,1—0,3 г. Необходимую величину навески выбирают исходя из норм осадков и зная относительное содержание определяемого элемента в веществе[11].
Навеска вещества на часовом стекле
Техника взятия навески может быть различной:
Часовое стекло или бюкс точно взвешивают, помещают туда приблизительно необходимое количество вещества и снова взвешивают. Массу вещества рассчитывают по разнице масс часового стекла или бюкса с навеской и без.
На часовое стекло или в бюкс помещают вещество в количестве, достаточном для взятия нескольких навесок, и взвешивают. Затем отсыпают порцию вещества в стакан, снова взвешивают часовое стекло или бюкс с остатками вещества и по разности результатов двух взвешиваний находят массу навески. Такой способ более удобен в тех случаях, когда необходимо взять несколько навесок анализируемого вещества[12].
Полученную таким образом навеску необходимо растворить. Как правило, для растворения используют минеральные кислоты. Иногда используют растворы щелочей или смеси кислот (например, царскую водку). В некоторых случаях для перевода определяемого элемента в растворимое соединение применяют сплавление с различными плавнями, часто карбонатами и нитратами щелочных металлов[13][14].
Перед проведением анализа необходимо подготовить раствор. Это подразумевает создание требуемого pH, маскировку мешающих ионов путём их осаждения или связывания в прочные комплексы, при необходимости упаривание раствора
1.3. Осаждение
Следующим этапом анализа является осаждение определяемого элемента. К осаждаемой форме предъявляются следующие требования[16]:
Малая растворимость: потеря массы осадка не должна превышать погрешность аналитических весов (0,1 мг).
Осадок должен быть чистым, то есть как можно меньше сорбировать посторонние ионы. По этой причине более предпочтительны кристаллические осадки, чем аморфные.
Осаждаемая форма должна легко переходить в гравиметрическую.
Образовываться в форме, удобной для последующих операций.
Процесс осаждения отличается в зависимости от характера осадка. Крупнокристаллические осадки получаются более чистыми, поэтому для снижения скорости образования центров кристаллизации[17] их получают путём осаждения из горячих разбавленных растворов, медленно (по каплям) приливая раствор осадителя, при перемешивании стеклянной палочкой[18]. После осаждения кристаллические осадки некоторое время (от 30 минут до 20 часов) выдерживают под маточным раствором[19]. Аморфные осадки осаждают из концентрированных горячих растворов. После осаждения к ним добавляют около 100 мл горячей воды, перемешивают и фильтруют сразу, не выдерживая под маточным раствором[20].
1.4. Фильтрование и промывание осадка
Для фильтрования используют беззольные фильтры, то есть такие, массой золы которых можно пренебречь. Крупнокристаллические осадки промывают непосредственно на фильтре, мелкокристаллические и аморфные — декантацией. При декантации раствор сливают через фильтр, стараясь оставить осадок в стакане. Затем к осадку добавляют новую порцию промывной жидкости, перемешивают, дают снова осесть и повторяют эту операцию несколько раз. После нескольких циклов осадок количественно переносят на фильтр. В качестве промывной жидкости может использоваться дистиллированная вода, однако чаще для уменьшения потерь осадка при фильтровании применяют разбавленный раствор осадителя
1.5. Высушивание и прокаливание
После фильтрования и промывания осадок переводят в гравиметрическую форму, к которой предъявляются следующие требования:
- точное соответствие состава химической формуле;
- устойчивость на воздухе (например, гравиметрическая форма не должна поглощать влагу или CO2);
- достаточно большая молекулярная масса и малая массовая доля определяемого элемента (для снижения погрешности).
Для получения гравиметрической формы осадок высушивают и/или прокаливают, в зависимости от вещества. Как правило, осадки, образованные органическим осадителем, только высушивают, а неорганические осадки прокаливают. Некоторые вещества при прокаливании претерпевают химические изменения (например, гидроксид железа(III) переходит в оксид), другие (такие как сульфат бария BaSO4) — нет[22].
![{\displaystyle {\ce {2 Fe(OH)3 ->[t] Fe2O3 + 3 H2O}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5013b9b5bc6c6e47458d5112cc723089b9f6d346)
Высушивание проводят в сушильном шкафу при температуре 90—105 °С или на воздухе, если не требуется высокая скорость. После высушивания фильтр озоляют, то есть нагревают в присутствии кислорода, сначала обугливая его, а затем окисляя (продукты окисления — CO и CO2 — газы), но без открытого горения. Некоторые вещества восстанавливаются углём, поэтому с ними работают иначе. Например, хлорид серебра счищают с фильтра, фильтр озоляют, после чего восстановленное серебро в тигле окисляют царской водкой вновь до хлорида серебра, а избыток кислот выпаривают, после чего переносят основную массу осадка обратно в тигель.
Прокаливание проводят в муфельных (или тигельных) печах, в тиглях, доведённых до постоянной массы. После прокаливания тигли помещают в эксикатор для предотвращения поглощения влаги из воздуха
1.6. Взвешивание и расчёт результатов анализа
Взвешивание производят на аналитических весах. Как правило, погрешность взвешивания составляет 0,1 мг. По известной массе гравиметрической формы нетрудно вычислить массу определяемого иона. Например, при определении бария, когда гравиметрической формой является сульфат бария BaSO4, массу бария можно вычислить по формуле

Отношение молярной массы определяемого вещества (элемента) к молярной массе гравиметрической формы, с учётом стехиометрических коэффициентов, называют фактором пересчёта и обозначают буквой [24].

Массовую долю определяемого иона в навеске (и, соответственно, в пробе) можно вычислить по формуле:

1.7. Растворимость осадков и факторы, влияющие на неё
Постоянной величиной, характеризующей растворимость вещества, является его произведение растворимости. Однако у одного и того-же вещества в разных условиях растворимость, т. е. концентрация насыщенного раствора, может отличаться. Для того, чтобы добиться полноты осаждения, но при этом поддерживать невысоким относительное пересыщение[25] для роста крупных кристаллов, необходимо учитывать факторы, влияющие на растворимость осадков.
Избыток одноимённых ионов
Для более полного осаждения определяемого элемента используют избыток ионов осадителя. Например, при осаждении свинца в виде PbSO4 для осаждения применяют избыток сульфат-ионов. Произведение растворимости сульфата свинца постоянно:
![{\displaystyle K_{s}({\text{PbSO4}})=[{\text{Pb}}^{2+}]\cdot [{\text{SO}}_{4}^{2-}]=1{,}6\cdot 10^{-8}{\frac {{\text{mol}}^{2}}{{\text{L}}^{2}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/35915c20c2a41ba66524ca3056259c9b579be73f)
Из уравнения видно, что при увеличении концентрации сульфат-ионов, концентрация ионов свинца будет понижаться.
При осаждении, как правило, не используют слишком большой избыток осадителя, поскольку в некоторых случаях при его увеличении возможно увеличение растворимости осадка из-за других эффектов, таких как комплексообразование[26].
Влияние pH (гидролиз)
Часто осадки являются солями слабых кислот, которые способны подвергаться гидролизу. Рассмотрим этот эффект на примере оксалата кальция:



В таком случае при подкислении раствора концентрация ионов C2O42- будет понижаться, следовательно, растворимость осадка будет повышаться[27].
Стоит отметить, что понижение pH может способствовать протонированию многих лигандов, разрушению комплексов, образованных ими, и привести к выпадению в осадок замаскированных ионов. Например, при подкислении раствора, содержащего хлорид-ионы и замаскированные аммиаком ионы серебра, хлорид серебра выпадет в осадок[28]:
![{\displaystyle {\ce {[Ag(NH3)2]+ + 2H+ <=> Ag+ + 2 NH4+}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/aee9ad3693d5d7d022fc535a91fcb681cf04af83)

Комплексообразование
На растворимость существенное влияние оказывает комплексообразование. Катионы многих металлов способны образовывать комплексные соединения, в результате чего их концентрация в свободном виде может существенно снижаться. Иногда этим эффектом пользуются, для того, чтобы замаскировать ионы, мешающие проведению анализа, тогда это называют маскировкой. В других случаях растворимость осадка нежелательна, и посторонние комплексообразователи, если они присутствуют, требуется удалить (например, органические кислоты окисляют до CO2 и воды).
В качестве лигандов могут выступать как сторонние ионы или молекулы, так и ионы, входящие в состав осадка. Как правило, в таких случаях при небольшом их избытке растворимость уменьшается, а затем увеличивается, поэтому часто для осаждения применяют 50%-ный избыток осадителя. Однако так происходит не всегда: например, даже небольшой избыток осадителя увеличивает растворимость HgI2[29].
Температура
Зависимость произведения растворимости от температуры количественно описывается формулой
- ,
 ,
,где:
- — произведение растворимости,
- — температура,
- — изменение энтальпии (тепловой эффект) реакции растворения,
- — универсальная газовая постоянная.
 —
—  — температура,
— температура, — изменение
— изменение  —
— Для растворения большинства малорастворимых соединений, требуется затратить энергию (), поэтому их растворимость с ростом температуры увеличивается[30].
 ), поэтому их растворимость с ростом температуры увеличивается
), поэтому их растворимость с ростом температуры увеличиваетсяИногда от температуры зависит и состав осадка. Например, осадок сульфата кальция при температурах до 60 °C имеет состав , но при более высокой температуре он переходит в . Поскольку растворение последнего экзотермично, растворимость осадка достигает максимума при 60 °C[31].
 , но при более высокой температуре он переходит в
, но при более высокой температуре он переходит в  . Поскольку растворение последнего экзотермично, растворимость осадка достигает максимума при 60 °C
. Поскольку растворение последнего экзотермично, растворимость осадка достигает максимума при 60 °CСолевой эффект
В расчётах произведение растворимости, т. е. произведение концентраций ионов, составляющих вещество, в соответствующих степенях, часто считают постоянным. На самом деле, более точно считать постоянным произведение не молярных концентраций, а активностей, т. е. концентраций, домноженных на коэффициенты активности. Коэффициенты активности зависят от ионной силы раствора, на которую влияет присутствие посторонних электролитов. Так например, растворимость PbSO4 в 0,1 M растворе KNO3 примерно в 3 раза выше, чем в чистой воде[32].
2. Титриметрический анализ
Титриметрический анализ (титрование) — метод количественного/массового анализа, который часто используется в аналитической химии, основанный на измерении объёма раствора реактива точно известной концентрации, расходуемого для реакции с определяемым веществом. Титрование — процесс определения титра исследуемого вещества. Титрование производят с помощью бюретки, заполненной титрантом до нулевой отметки. Титровать начиная от других отметок не рекомендуется, так как шкала бюретки может быть неравномерной. Заполнение бюреток рабочим раствором производят через воронку или с помощью специальных приспособлений, если бюретка полуавтоматическая. Конечную точку титрования (не следует путать с точкой эквивалентности) определяют с помощью индикаторов или физико-химическими методами (по электропроводности, светопропусканию, потенциалу индикаторного электрода и т. д.). По количеству затраченного на титрование рабочего раствора рассчитывают результаты анализа.
3. Термический анализ
Термический анализ представляет собой метод исследования физико-химических и химических превращений, происходящих в веществе при программированном изменении температуры как при нагревании, так и при охлаждении.
Родоначальник термического анализа французский физик и химик (фр. Henri Louis Le Chatelier, годы жизни 8 октября 1850 — 17 сентября 1936).

С помощью этого метода обнаруживают тепловую природу, эндо- или экзотермический характер и температурный интервал превращения
В процессе нагревания или охлаждения вещества, регистрируются не только его тепловые свойства, но и изменения массы, объема, состава и количества выделяющихся газов, электропроводности, магнитной восприимчивости и т.д. термические методы делятся:
Классификация термических методов анализа
|
Название |
Принцип метода |
|
Дифференциальный термический анализ(ДТА) |
Основан на регистрации тепловых эффектов,сопровождающих физические превращения и химические реакции, происходящие под воздействием высоких температур. |
|
Термогравиметрия (ТГ) |
Основан на получении и изучении закономерностей изменения веса вещества при нагревании. |
|
Термодилатометрия |
Позволяет определять изменение длины и объема образца в зависимости от температуры. |
|
Термомагнитометрия |
Изучение магнитных свойств минералов в зависимости от температуры
|
|
Термоволюметрия |
Позволяет регистрировать объем выделяющегося из образца газов при повышении температуры. |
Результатом термического анализа являются термические кривые - термограммы (кривые нагревания), которые зависят главным образом от химического состава и структуры исследуемого вещества.
С помощью термогравиметрии исследуют:
- Термодеструкцию материалов
- Содержание примесей
- Содержание растворителей и воды
- Сложные многокомпонентные смеси
- Термостойкость и др.
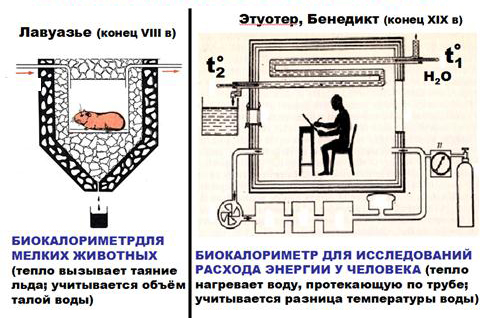
Калориметрия (от лат. calor — тепло и лат. metro — измеряю) — совокупность методов измерения количества теплоты, выделяющейся или поглощаемой при протекании различных физических или химических процессов. Основателем калориметрии можно считать шотландского химика и физика (англ. Joseph Black, годы жизни 16 апреля 1728 — 6 декабря 1799).(материал из Wikipedia® )
Прямая калориметрия – непосредственный учет количества тепла, выделяемого организмом в биокалориметрах (камерах Лавуазье- Лапласа и Этуотера-Бенедикта), грубо говоря определение количества тепла, которое организм выделяет за сутки.
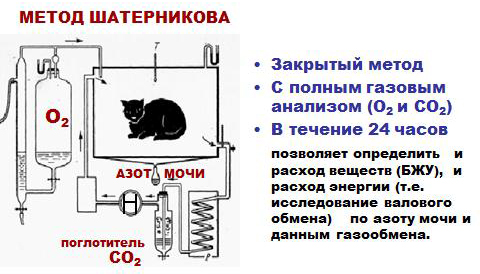
Непрямая калориметрия – определение теплообразования в организме по его газообмену – учет количества потребляемого кислорода и выделяемого углекислого газа с последующим расчетом основного обмена организма (способ Дуглас-Холдена, оксиспирография).

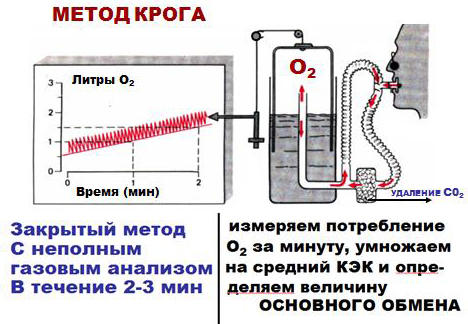
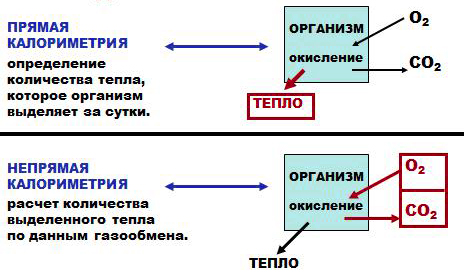
ДЫХАТЕЛЬНЫЙ КОЭФФИЦИЕНТ - Отношение
количества выделенного углекислого газа к количеству поглощенного кислорода.
ДК=СО2/О2
ДК зависит от характера пищи
ДКдля белков =0,8
ДК для углеводов =1,0
ДК для жиров =0,7
ДК при смешанной пище =0,85
С помощью дифференциальной сканирующей калориметрии (ДСК)
можно измерить характеристические температуры и выделяемое или поглощаемое
тепло физических процессов или химических реакций, происходящих в образцах
твердых тел и жидкостей при их контролируемом нагреве или охлаждении. ДСК
является наиболее часто используемым методом в термическом анализе.

Области применения:
Химическая и фармацевтическая промышленность, научные исследования
- Процессы с участием химически активных газов, таких как O2, H2 и CO2, а также горючих и токсичных газов
- Безопасные исследования под давлением
- Подавление процессов испарения (за счет повышения температуры кипения)
- Разделение химических реакций и процессов испарения, которые протекают одновременно при нормальном давлении
- Исследование реакций с участием летучих компонентов
- Исследование каталитических реакций
- Исследование гетерогенных реакций
- Исследование процессов адсорбции и десорбции
- Измерение зависимости давления от температуры кипения
- Определение энтальпии парообразования
Закон теплообмена Ньютона
q = a S (Тср – T),
где a – коэффициент теплообмена, Вт/м2·К,
S – площадь поверхности, через которую осуществляется теплообмен, м2,
Т – температура образца, К,
Тср – температура среды, окружающей образец (температура печи), К.
Тср = Т0 + v t,
где Т0 – начальная температура, К,
v º dТср/dt = const – скорость нагрева (охлаждения), К/с
Тепловой поток, передаваемый образцу при нагреве в процессе термического анализа
q = mCv [1– exp (– t / t)],
где
t = mC/(aS) – постоянная времени, характеризующая термическую инерцию образца.
Тепловой поток при фазовом превращении
qфп = – DH m (ρ/ρ0) dy/dt,
где DH – удельная теплота (энтальпия) превращения,
y º (V/V0) – объёмная доля новой фазы,
m и V0 – масса и начальный объём образца,
ρ0 и ρ – исходная плотность и плотность новой фазы.
Знак «–» введён для случая выделения тепла при фазовом превращении (как, например, в случае кристаллизации).
Форма кривой охлаждения согласно теории термического анализа
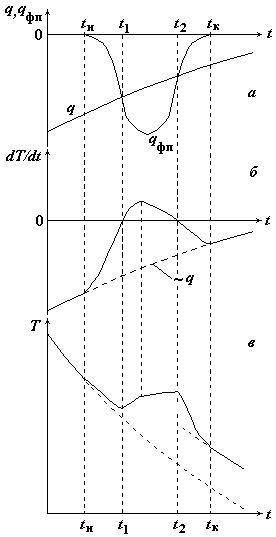
а – зависимости от времени теплового потока, отводимого от образца (q), и скорости выделения тепла в образце из-за фазового превращения (qфп);
б – временная зависимость скорости изменения температуры образца;
в – кривая термического анализа при охлаждении
Методика обработки кривых термического анализа
Интерпретация кривой термического анализа основана на изучении наклона кривой:
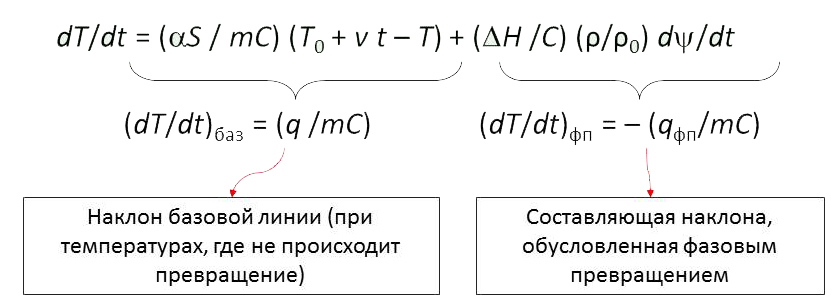
Поиск температур начала и конца превращения проводится путем построения касательных к кривой термического анализа и нахождения точки, где наклон касательных резко изменяется.
Закон охлаждения Ньютона в калориметре
q = αA(Tоб – T)
α – коэффициент теплообмена, Вт/м2·К.
Условия применимости на примере калориметра смешения:
1.Разность температур калориметрической системы и оболочки не должна превышать 2–3 градуса: (Tоб – T) < 2…3 К.
2. Внешняя поверхность калориметра и внутренняя поверхность оболочки должны обладать хорошей отражательной способностью.
3.Расстояние между стенками калориметрического сосуда и оболочки должно составлять около 10 мм; при том конвекция практически исключается, а тепловые потери из-за теплопроводности воздуха невелики.
4. Подъём температуры в опыте не должен превышать 2 К.
В методах смешения нагретый образец вводят в калориметр, температура которого повышается. Количество теплоты, введённое в калориметр:
Q = Cкал·(Tf – T0),
где Cкал – теплоёмкость калориметра (тепловой, или энергетический, эквивалент),
Tf и T0 – конечная и начальная температуры калориметрической системы (калориметрического вещества – не путать с температурой образца!).
Термин "микрокалориметрия'' неразрывно связано с именем Э. Кальве, который существенно усовершенствовал первые калориметры Тиана, основанные на измерении потока энергии, и разработал теорию метода для измерения малых количеств энергии.
Особенности калориметра Кальве
- Предназначен для измерения очень малых количеств
энергии и для исследования очень медленных процессов.
- Тепло отводится от калориметрической ячейки к
оболочке через термопары.
- Для повышения чувствительности измерений
используются термобатареи, состоящие из многих десятков и даже сотен
термопар.
- Измеряемый тепловой поток частично компенсируется
эффектом Пельтье.
Теория метода микрокалориметрии основывается на уравнение Тиана.
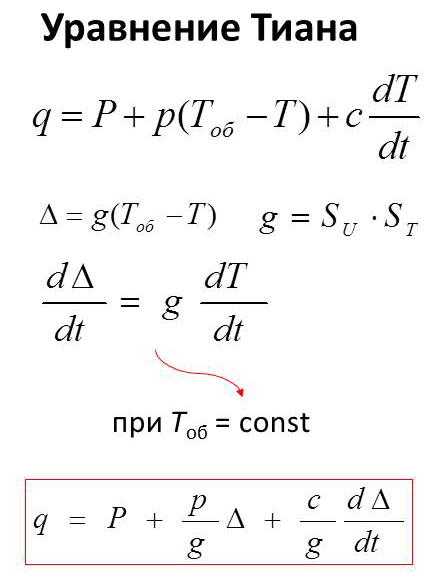
q – тепловой поток в калориметрической ячейке (количество энергии, выделяемое в калориметрической ячейке в единицу времени), Вт;
Р – та часть теплового потока (тепловой мощности), которая скомпенсирована эффектом Пельтье, Вт;
р = αS – количество энергии, теряемое калориметрической ячейкой в единицу времени при разности температур (Тоб – Т), равной 1 оС, Вт/К;
с – теплоёмкость (энергетический эквивалент) калориметрической ячейки, Дж/К,
Δ – отклонение гальванометра,
SU и Sт – чувствительности гальванометра и термобатареи.
4. Вискозиметрия
Вязкость, внутреннее трение - свойство жидкостей и газов оказывать сопротивление перемещению одного их слоя относительно другого.
Количественно вязкость характеризуется значением величины, называемой динамической вязкостью или коэффициентом внутреннего трения и обозначаемой n или u. Характерной особенностью этого вида трения является то, что оно наблюдается не на границе твердого тела и жидкости, а во всем объеме жидкости.
Единицей динамической вязкости в Международной системе единиц (СИ) является паскаль-секунда (Па*с). Паскаль-секунда равна динамической вязкости среды, касательное напряжение в которой при ламинарном (упорядоченном) течении и при разности скоростей слоев, находящихся на расстоянии 1 м по нормали к направлению скорости, равной 1 м/с, составляет 1 Па.
Кинематическая вязкость равна отношению динамической вязкости среды к ее плотности при той же температуре:
ν=η/ρ
Единицей кинематической вязкости в СИ является квадратный метр в секунду (м2/с). При кинематической вязкости 1 м2/с динамическая вязкость среды плотностью 1 кг/м3 равна 1 Па*с.
Типы вискозиметров
В зависимости от способа измерения вискозиметры подразделяются на:
- капиллярные (вискозиметры истечения),
- шариковые, ротационные,
- вибрационные
- ультразвуковые.
При пользовании капиллярными вискозиметрами измеряется время истечения известного количества (объема) жидкости сквозь капиллярные трубки определенного диаметра. Стеклянные капиллярные вискозиметры чаще других используются в практике химических лабораторий.
Капиллярный вискозиметр представляет собой один или несколько резервуаров заданного объёма с отходящими трубками малого круглого сечения, или капиллярами. Принцип действия капиллярного вискозиметра заключается в медленном истечении жидкости из резервуара через капилляр определенного сечения и длины под влиянием разности давлений. Суть опыта при определении вязкости состоит в измерении времени протекания известного количества жидкости при известном перепаде давлений на концах капилляра.


- материал - боросиликатное стекло;
- наличие встроенного уплотнителя;
- точность, % - ± 0,3%;
Вискозиметр с падающим шариком. Использует для определения вязкости жидкости, измеряя время, требуемое шарику для падения под собственным весом через трубку, заполненную жидким образцом.


- Диапазон вязкости, мПа×с — 0,5 – 100 000;
- температурный диапазон, °С – от -20 до +120;
- точность, % — ±1;
- сходимость, % — 0,5;
- материал шариков 1, 2 и G - боросиликатное стекло, шариков 3 и 4 - железоникелевый сплав, шариков 5 и 6 – нержавеющая сталь.
Ротационный вискозиметр. Прибор позволяет измерить реологические свойства различных материалов в вакууме, инертных газовых средах а широком интервала температур.


- Диапазон вязкости, мПа×с — 20 – 2 000 000;
- количество скоростей – 18;
- диапазон скоростей, об/мин - 0,3 – 100;
- точность, % — ±1;
- сходимость, % — 0,2;
- ЖК-дисплей, сенсорная цифровая клавиатура;
- отображение установленной скорости и текущего значения вязкости в сПз (мПа*с), температуры исследуемого образца (опция);
- определение скорости сдвига (при использовании коаксиального шпинделя) SR (сек-1);
- определение касательного напряжения (при использовании коаксиального шпинделя) SS (Н/м2);
- определение динамической (сПз или мПа·с) или кинематической (cСт) вязкостей (при вводе данных о плотности образца);
- предустановка необходимого вращающего момента;
- функция автоматического отключения с таймером;
- 10 программ памяти;
- меню на 10 языках;
- функция самотестирования с сигнализацией неисправности Auto-Test;
- функция автоматической установки диапазона измерений Auto-Range;
- интерфейс USB;
Вискозиметр вибрационный. Принцип действия основан на определении изменений параметров вынужденных колебаний тела правильной геометрической формы, называемого зондом вибрационного вискозиметра, при погружении его в исследуемую среду.


- Возможность непрерывного измерения вязкости;
- высокая точность измерения;
- широкий диапазон измерений без замены сенсорных пластин;
- измерение вязкости жидкостей, содержащих пузырьки;
- измерение вязкости взвесей и текучего образца;
- выносной вакуум-флюоресцентный дисплей;
- возможность подключения к ПК через стандартный интерфейс RS-232С и стандартный USB интерфейс;
- частота вибрации, Гц — 30;
- диапазон измерения, мПа х с — 0,3 — 1 000;
- относительная погрешность измерения, % — ±5;
- минимальный размер образца, мл — 2;
- единицы измерения — мПа х с, Па х с, сП, П.
5. Референтная методика измерений
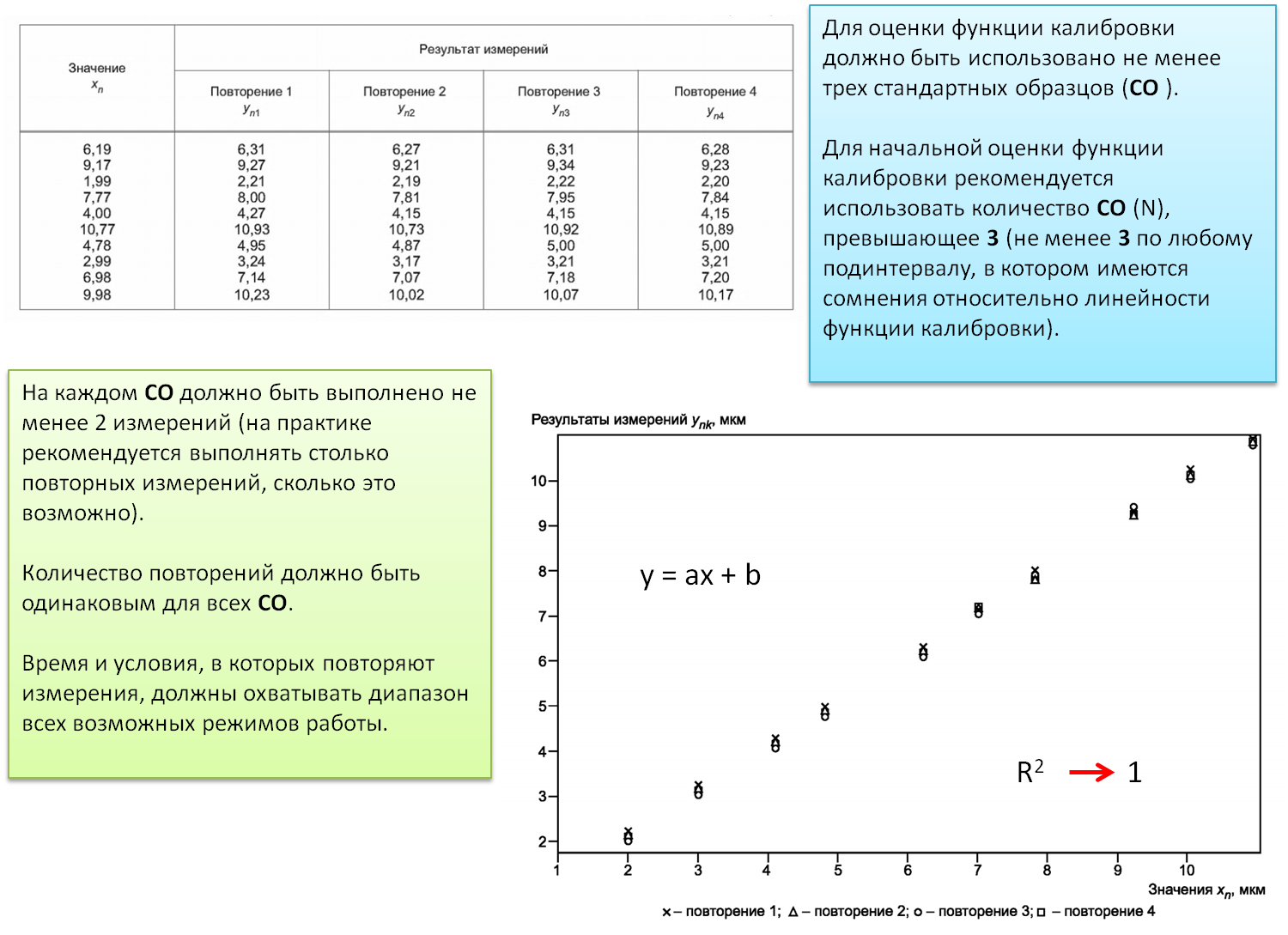
Референтная методика измерений: Методика измерений, принятая для получения результатов измерений, которые могут быть использованы для оценки правильности измеренных значений величины, полученных по другим методикам измерений величин того же рода, а также для калибровки или для определения характеристик стандартных образцов.
Все средства измерений, используемые для испытаний и/или калибровочных работ, включая средства для вспомогательных измерений (например, для контроля параметров окружающей среды), имеющих значительное влияние на точность и достоверность результатов испытания, калибровки или отбора образцов, должны быть калиброваны перед вводом в эксплуатацию.В лаборатории должны быть установлены программа и процедура проведения калибровки средств измерений.
Примечание - Такая программа должна включать в себя систему выбора, использования, калибровки, проверки, контроля и поддержания эталонов единиц физических величин, стандартных образцов, применяемых в качестве эталонов, а также измерительного и испытательного оборудования, используемого при проведении испытаний и калибровки.
ГОСТ ИСО/МЭК 17025-2009. Общие требования к компетентности испытательных и калибровочных лабораторий
ГОСТ Р ИСО 11095-2007 Статистические методы. Линейная калибровка с использованием образцов сравнения