Молекулярные мишени гиполипидемических средств
| Сайт: | Электронный информационно-образовательный портал ВолгГМУ |
| Курс: | Медицинская биохимия (10 семестр) |
| Книга: | Молекулярные мишени гиполипидемических средств |
| Напечатано:: | Гость |
| Дата: | Четверг, 30 октября 2025, 00:41 |
1. Введение
Среди нарушений обмена веществ одно из лидирующих мест занимают нарушения обмена липидов, или дислипидемии. Термин «дислипидемии» (дислипопротеинемии) включает в себя наследственные или приобретенные состояния, характеризующиеся нарушением синтеза, катаболизма, удаления из циркуляции липидов и липопротеинов, что приводит к нарушению их содержания в крови (повышению или понижению). В сравнении с термином «дислипопротеинемия» более широким по охвату нарушений уровня липопротеинов в крови считается термин «гиперлипопротеинемия», который отражает повышенное содержание в крови одного или нескольких классов липопротеинов.
Клиническими проявлениями дислипидемии являются ксантомы – плотные узелки, содержащие холестерин, например, на кисти, подошвах ног, ладонях, коже любого участка тела, особенно спины, а также ксантелазмы – отложение холестерина под кожей век в виде плоских узелков желтого цвета или узелков, не отличающихся по цвету от других участков кожи. При дислипидемии наблюдаются атеросклеротические поражения органов. Атеросклероз – хроническое заболевание, характеризующееся поражением артерий эластического и мышечного типа в виде очаговых отложений липидов и белков в интиме сосудов и реактивной клеточной реакции их стенки, что ведет к сужению просвета сосудов и нарушению физиологических функций пораженных артерий и, как следствие, к расстройствам системы кровообращения.
Сегодня в лечении дислипидемии используется 5 основных классов лекарственных средств (ЛС), применяемых с учетом механизма их действия, эффективности и возможности появления нежелательных эффектов: статины; никотиновая кислота и ее производные; фибраты; секвестранты желчных кислот; антиоксиданты.
2. Статины
Среди этих ЛС особого внимания заслуживают статины, появление которых стало возможным благодаря открытию японского ученого Akira Endo, выделившего в 1971 г. из грибов рода Penicillium компонент под названием «компактин», ставший родоначальником статинов. Это соединение обладало способностью снижать уровень холестерина.

С тех пор прошло достаточное количество времени, прежде чем на фармацевтическом рынке в 1987 г. появился первый коммерческий статин под названием «ловастатин» (Мевакор), выделенный из гриба рода Aspergillus. Механизм действия статинов реализуется через ингибирование фермента 3-гидрокси-3-метилглютарил-кофермента А редуктазы (ГМГ-КоА-редуктаза), что приводит к уменьшению уровня внутриклеточного холестерина с последующей активацией транскрипции гена рецепторов липопротеинов низкой плотности и, соответственно, снижению содержания ЛПНП в крови. Статины являются общепризнанным золотым стандартом в лечении гиперлипидемии. За 30 лет их применения они доказали свою эффективность и стали доминирующей группой препаратов, используемых при дислипидемиях.
Однако у каждого пятого человека, принимающего статины, по разным причинам не удается достичь целевых показателей липидов. Некоторые пациенты не переносят статины из-за развития побочных реакций (миалгия, миопатия, миозит, рабдомиолиз, увеличение риска повышения уровня глюкозы в крови и развитие сахарного диабета 2 типа, поражение печени). Кроме того, у значительного количества пациентов с высокой гиперхолестеринемией, в первую очередь семейной гиперхолестеринемией (СГ), даже высоких доз статинов и вспомогательной терапии недостаточно для достижения целевых уровней липидов (2,3). Гиперхолестеринемия – это повышение уровня общего холестерина в 2-2,5 раза по сравнению с нормой за счет ЛПНП. СГ – наиболее распространенное (и очень опасное) наследственное заболевание в мире, без своевременного лечения которого больным угрожают раннее и агрессивное развитие атеросклероза, стеноза аорты, ранние инфаркты, инсульты и наступление внезапной смерти. Как сказал J. Kastelein (4), без лечения у больных с СГ прогноз такой же плохой, как и у больных СПИД(ом). Гетерозиготная форма СГ в общей популяции в большинстве стран встречается довольно часто (от 1:200 до 1:500), в то время как гомозиготная форма проявляется намного реже (1:1 000 000 человек). Считается, что во всем мире больных СГ может быть от 20 до 34 млн, но выявлено заболевание не более чем у 10% из них, а адекватное лечение получают только 5%.
Учитывая важность проведения гиполипидемической терапии для больных с нарушениями липидного обмена, были разработаны ЛС с новыми, уникальными, механизмами действия. Эти ЛС отличаются от известных классов гиполипидемических средств, таких как статины, секвенаторы желчных кислот, ингибиторы абсорбции холестерина.
2.1. Аторвастатин - типичный статин (информация из РЛС)
Структурная формула

Русское название
АторвастатинЛатинское название вещества Аторвастатин
Atorvastatinum (род. Atorvastatini)Химическое название
[R(R*R*)]-2-(4-Фторфенил)-бета,дельта-дигидрокси-(1-метилэтил)-3-фенил-4- [(фениламино)карбонил]-пиррол-1-гептановая кислота (и в виде кальциевой соли)
Брутто-формула
C33H35FN2O5Фармакологическая группа вещества Аторвастатин
Нозологическая классификация (МКБ-10)
Код CAS
134523-00-5Характеристика вещества Аторвастатин
Гиполипидемическое средство из группы статинов. Ингибитор ГМГ-КоА-редуктазы.
Аторвастатин кальция — белый или почти белый кристаллический порошок, нерастворим в водных растворах со значениями pH 4 и ниже; незначительно растворим в дистиллированной воде, фосфатном буфере с pH 7,4 и ацетонитриле; мало растворим в этаноле, легко растворим в метаноле. Молекулярная масса 1209,42.
Фармакология
Фармакологическое действие - гиполипидемическое.Конкурентно селективно ингибирует ГМГ-КоА-редуктазу, превращающую 3- гидрокси −3-метилглутарил-КоА в мевалоновую кислоту (предшественник стеролов, включая холестерин). Триглицериды и холестерин в печени включаются в состав ЛПОНП, поступают в плазму и транспортируются в периферические ткани. Из ЛПОНП образуются ЛПНП, которые катаболизируются при взаимодействии с высокоаффинными рецепторами ЛПНП. Повышение плазменных уровней общего холестерина, холестерина ЛПНП, аполипопротеина В способствует развитию атеросклероза и является фактором риска развития сердечно-сосудистых заболеваний, тогда как повышение уровня ЛПВП ассоциировано со снижением риска сердечно-сосудистых осложнений.
Аторвастатин снижает уровни холестерина и липопротеинов в плазме крови, ингибируя ГМГ-КоА-редуктазу, а также тормозит синтез холестерина в печени, увеличивая число ЛПНП- рецепторов на поверхности клеток, способствует усилению захвата и катаболизма ЛПНП. Подавляет образование ЛПНП и число частиц ЛПНП. Уменьшает уровень холестерина-ЛПНПу больных гомозиготной семейной гиперхолестеринемией, которая обычно устойчива к терапии гиполипидемическими средствами.
У большинства пациентов действие проявляется через 2 нед после начала терапии, максимальный эффект развивается к 4-й неделе и сохраняется в течение всего периода лечения.
Токсическое влияние на ЦНС. Кровоизлияние в мозг наблюдалось у собак при применении аторвастатина в течение 3 мес в дозе 120 мг/кг/сут, кровоизлияние в мозг и образование вакуолей зрительного нерва — у самок собак после 11 мес применения аторвастатина в дозах до 280 мг/кг/сут. Доза 120 мг/кг/сут создавала системную экспозицию примерно в 16 раз превышающую таковую при назначении максимальной терапевтической дозы для человека (80 мг/сут) в пересчете на AUC (0–24 ч). В двухгодичном исследовании были зафиксированы одиночные тонические судороги у двух собак (у одной — при дозе 10 мг/сут, у другой — при дозе 120 мг/сут). Не отмечалось признаков повреждения ЦНС у мышей при хроническом введении аторвастатина в период вплоть до 2 лет при дозах до 400 мг/сут и у крыс при дозах до 100 мг/сут. Эти дозы создавали экспозицию, в 6 раз (мыши) и в 11 раз (крысы) превышающую экспозицию у человека при рекомендуемой терапевтической дозе 80 мг в пересчете на AUC(0–24 ч).
Сосудистые повреждения в ЦНС, характеризующиеся периваскулярными кровоизлияниями, отеком и мононуклеарно-клеточной инфильтрацией периваскулярных пространств, отмечались у собак, которым давали другие препараты этого класса. Химически сходные вещества того же класса, что и аторвастатин, вызывали дозозависимую дегенерацию зрительного нерва при дозе, создающей плазменную концентрацию, в 30 раз превышающую таковую у человека при приеме максимально рекомендуемой дозы.
Канцерогенность, мутагенность, влияние на фертильность
В двухгодичных исследованиях канцерогенности у крыс с использованием доз аторвастатина 10, 30 и 100 мг/кг/сут две редкие опухоли в мышцах были обнаружены у самок при максимальной дозе: в одном случае это была рабдомиосаркома, в другом — фибросаркома. Эта доза соответствовала значениям системной экспозиции (плазменная AUC0–24 ч), в 16 раз превышающим величины экспозиции у человека при пероральной дозе 80 мг. В двухгодичных исследованиях канцерогенности у мышей при дозах аторвастатина 100, 200 и 400 мг/кг/сут было отмечено значимое повышение частоты развития аденомы печени при максимальной дозе у самцов и карциномы печени при максимальной дозе у самок. Эти случаи наблюдались при значении плазменной AUC0 –24 ч, примерно в 6 раз превышающем величину экспозиции у человека при пероральной дозе 80 мг.
In vitro аторвастатин не проявлял мутагенных или кластогенных свойств в следующих тестах (с/без метаболической активации): тест Эймса с Salmonella typhimurium и Escherichia coli, тест с гипоксантин-гуанинфосфорибозилтрансферазой и тест на хромосомные аберрации на клетках яичника китайского хомячка. Не выявлено мутагенности in vivo в микроядерном тесте у мышей.
В исследованиях у крыс при дозах аторвастатина до 175 мг/кг (экспозиция в 15 раз выше экспозиции у человека) негативного влияния фертильность не выявлено. При введении крысам аторвастатина в дозе 100 мг/кг/сут (в 16 раз выше AUC у человека при дозе 80 мг) в течение 3 мес зафиксированы аплазия и аспермия в придатках яичка у 2 из 10 крыс; масса яичек была значительно снижена при дозах 30 и 100 мг/кг и масса придатков была ниже при дозе 100 мг/кг. У самцов крыс, которые получали аторвастатин в дозе 100 мг/кг/сут в течение 11 нед до спаривания, была снижена подвижность спермы и ее концентрация, а количество аномальных сперматозоидов повышено. Аторвастатин не оказывал неблагоприятного влияния на параметры спермы и репродуктивные органы у собак, получавших его в дозах 10, 40 и 120 мг/кг в течение 2 лет.
Аторвастатин быстро всасывается после приема внутрь, степень абсорбции увеличивается пропорционально повышению дозы, Cmax достигается в течение 1–2 ч. Абсолютная биодоступность — примерно 14%, системная доступность в отношении ингибирования ГМГ-КоА-редуктазы — примерно 30%. Низкая системная доступность обусловлена пресистемным метаболизмом в слизистой оболочке ЖКТ и/или при «первом прохождении» через печень. Средний объем распределения аторвастатина — примерно 381 л. Связывание с белками плазмы составляет ≥98%. Соотношение уровней кровь/плазма равно примерно 0,25, что свидетельствует о слабом проникновении аторвастатина в эритроциты. В печени аторвастатин экстенсивно метаболизируется при участии изофермента CYP3А4 цитохрома P450 с образованием орто- и парагидроксилированных производных и различных продуктов бета-окисления. In vitro орто- и парагидроксилированные метаболиты оказывают ингибирующее действие в отношении ГМГ-КоА-редуктазы, сопоставимое с таковым аторвастатина. Ингибирующий эффект в отношении ГМГ-КоА-редуктазы примерно на 70% определяется активностью циркулирующих метаболитов. Аторвастатин и его метаболиты выводятся главным образом с желчью после печеночного и/или внепеченочного метаболизма (не подвергается выраженной кишечно- печеночной рециркуляции). Т1/2 аторвастатина — 14 ч. Благодаря наличию активных метаболитов ингибирующая активность в отношении ГМГ-КоА-редуктазы сохраняется около 20 –30 ч. Менее 2% принятой внутрь дозы аторвастатина определяется в моче. Не выводится в ходе гемодиализа.
Особенности фармакокинетики у отдельных групп пациентов
У пожилых людей плазменные концентрации аторвастатина выше (примерно 40% для Cmax и 30% для AUC), чем у взрослых пациентов молодого возраста. Значения Cmax у женщин выше на 20%, AUC — ниже на 10% (клинического значения не имеет). Заболевания почек не влияют на плазменные концентрации аторвастатина или его эффект на показатели липидного обмена. У больных алкогольным циррозом печени концентрации аторвастатина значительно повышены: при нарушении функции печени легкой степени выраженности (Child Pugh Class A) — в 4 раза (и C max, и AUC), при нарушении функции средней степени выраженности (Child Pugh Class B) — Cmax в 16 раз, AUC — в 11 раз выше нормы.
Применение вещества Аторвастатин
Первичная гиперхолестеринемия (гетерозиготная семейная и несемейная гиперхолестеринемия, по Фредриксону тип IIa), комбинированная (смешанная) гиперлипидемия (по Фредриксону типы IIb и III), дисбеталипопротеинемия (по Фредриксону тип III) (в качестве дополнения к диете), семейная эндогенная гипертриглицеридемия (по Фредриксону тип IV), резистентная к диетическим методам лечения. Гомозиготная наследственная гиперхолестеринемия (в качестве дополнения к гиполипидемической терапии, в т.ч. аутогемотрансфузии очищенной от ЛПНП крови). Заболевания сердечно-сосудистой системы (в т.ч. у пациентов без клинических проявлений ИБС, но имеющих повышенные факторы риска ее возникновения — возраст старше 55 лет, никотиновая зависимость, артериальная гипертензия, генетическая предрасположенность), в т.ч. на фоне дислипидемии — вторичная профилактика с целью снижения суммарного риска смерти, инфаркта миокарда, инсульта, повторной госпитализации по поводу стенокардии и необходимости в реваскуляризации.
Противопоказания
Гиперчувствительность, заболевания печени в активной стадии (в т.ч. активный хронический гепатит, хронический алкогольный гепатит), повышение активности печеночных трансаминаз (более чем в 3 раза по сравнению с верхней границей нормы) неясного генеза, печеночная недостаточность, цирроз печени любой этиологии, беременность и период лактации.
Ограничения к применению
Заболевания печени в анамнезе, тяжелые нарушения электролитного баланса, эндокринные и метаболические нарушения, алкоголизм, артериальная гипотензия, тяжелые острые инфекции (сепсис), неконтролируемые судороги, обширные хирургические вмешательства, травмы, возраст до 18 лет (эффективность и безопасность не установлены).
Применение при беременности и кормлении грудью
Аторвастатин проходит через плаценту и достигает в печени плода уровня, эквивалентного уровню в плазме матери. Аторвастатин не проявлял тератогенности у крыс при использовании в дозах до 300 мг/кг/сут и у кроликов при дозах до 100 мг/кг/сут. Эти дозы создавали экспозицию, в 30 (крысы) и 20 (кролики) раз превышающую экспозицию у человека (в пересчете на площадь поверхности тела, в мг/м2).
В исследованиях у крыс, получавших аторвастатин в дозах 20, 100 и 225 мг/кг/сут с 7-го дня беременности до 21 дня лактации, отмечалось снижение выживаемости детенышей при рождении, новорожденных, и созревание детенышей самок, получавших дозы аторвастатина 225 мг/кг/сут. Зафиксировано снижение массы тела на 4 и 21 день у детенышей самок, получавших аторвастатин в дозе 100 мг/кг/сут; снижение массы тела при рождении, на 4, 21 и 91 день — при дозе 225 мг/кг/сут. Задержка развития отмечалась при дозе 100 мг/кг/сут(роторная активность) и 225 мг/кг/сут (испуг при звуках, нарушение формирования ушной раковины, время открытия глаз). Эти дозы соответствуют величинам AUC, в 6 (100 мг/кг) и 22 раза (225 мг/кг) превышающим AUC у человека при дозе 80 мг/сут. Редкие случаи врожденных аномалий наблюдались после внутриутробной экспозиции ингибиторов ГМГ-КоА-редуктазы.
Холестерин и другие вещества, синтезируемые из холестерина, важны для развития плода (включая синтез стероидов и клеточных мембран). Поскольку ингибиторы ГМГ-КоА-редуктазы снижают синтез холестерина и, возможно, синтез других биологически активных веществ — производных холестерина, эти ЛС могут оказывать вредное эмбриональное воздействие при приеме беременными женщинами. В связи с этим ингибиторы ГМГ-КоА-редуктазы противопоказаны в период беременности и грудного вскармливания.
Имеется одно сообщение о тяжелой врожденной костной деформации, трахео-эзофагеальном свище и анальной атрезии (VATER-ассоциация) у ребенка, родившегося у матери, принимавшей ловастатин с декстроамфетамина сульфатом в I триместре беременности.
Безопасность применения аторвастатина у беременных женщин не установлена.
Категория действия на плод по FDA — Х.
Женщинам детородного возраста аторвастатин можно принимать только в случае использования ими надежных мер контрацепции. Если больная планирует беременность, она должна прекратить прием препарата по крайней мере за 1 мес до запланированной беременности. В случае наступления беременности в период лечения прием аторвастатина следует немедленно прекратить. Пациентка должна быть проинформирована о возможном риске для плода.
В экспериментах на животных установлено, что аторвастатин проникает в грудное молоко крыс. Уровни ЛС в плазме и печени детенышей кормящих животных составляют от таковых в материнском молоке 50 и 40% соответственно.
Неизвестно, секретируется ли аторвастатин в грудное молоко у человека. Поскольку возможно серьезное негативное влияние на младенца, при приеме аторвастатина необходимо прекратить грудное вскармливание.
Побочные действия вещества Аторвастатин
В контролируемых клинических испытаниях (n=2502) менее 2% пациентов прекратили лечение в связи с побочными эффектами, вызванными аторвастатином. Наиболее частыми неблагоприятными эффектами, связанными с приемом аторвастатина, были запор, метеоризм, диспепсия и боль в животе.
Со стороны нервной системы и органов чувств: ≥2% — головная боль, астенический синдром, инсомния, головокружение; <2% — недомогание, сонливость, кошмарные сновидения, амнезия, парестезии, периферическая нейропатия, эмоциональная лабильность, нарушение координации движений, паралич лицевого нерва, гиперкинез, депрессия, гиперестезия, амблиопия, сухость конъюнктивы, нарушение аккомодации глаза, кровоизлияние в глаз, глаукома, шум в yшах, глухота, паросмия, потеря вкусовых ощущений, извращение вкуса.
Со стороны сердечно-сосудистой системы: ≥2% — боль в груди; <2% — сердцебиение, вазодилатация, обморок, мигрень, постуральная гипотензия, повышение АД, флебит, аритмия, стенокардия, анемия, лимфаденопатия, тромбоцитопения.
Со стороны респираторной системы: ≥2% — синусит, фарингит, бронхит, ринит; <2% — пневмония, диспноэ, бронхиальная астма, носовое кровотечение.
Со стороны органов ЖКТ: ≥2% — боль в животе, запор или диарея, диспепсия, метеоризм, тошнота; <2% — анорексия или повышение аппетита, сухость во рту, отрыжка, дисфагия, рвота, стоматит, эзофагит, эрозивно-язвенные поражения слизистой оболочки полости рта, гастроэнтерит, гастрит, энтерит, колит, хейлит, язва двенадцатиперстной кишки, язва желудка, панкреатит, желчная колика, холестатическая желтуха, нарушение функции печени, гепатит, ректальное кровотечение, мелена, кровоточивость десен, тенезмы.
Со стороны опорно-двигательного аппарата: ≥2% — артралгия, миалгия, артрит; <2% — ригидность мышц шеи, судороги мышц ног, бурсит, тендосиновит, миастения, миозит, кривошея, мышечный гипертонус, контрактуры суставов.
Со стороны мочеполовой системы: ≥2% — урогенитальные инфекции, периферические отеки; <2% — гематурия, альбуминурия, учащение мочеиспускания, цистит, дизурия, никтурия, нефролитиаз, недержание мочи или задержка мочеиспускания, императивные позывы на мочеиспускание, нефрит, вагинальное кровотечение, маточное кровотечение, метроррагия, эпидидимит, снижение либидо, импотенция, нарушение эякуляции.
Со стороны кожных покровов: <2% — алопеция, ксеродермия, повышенное потоотделение, акне, экзема, себорея, язвы кожи, экхимозы, петехии.
Аллергические реакции: ≥2% — кожная сыпь; <2% — отек лица, генерализованный отек, кожный зуд, контактный дерматит, крапивница.
Прочие: ≥2% — инфекции, случайная травма, гриппоподобный синдром, боль в спине; <2% — повышенная температура тела, фотосенсибилизация, повышение массы тела, увеличение молочных желез, гипергликемия, гипогликемия, повышение сывороточной креатинфосфокиназы, ЩФ, повышение АЛТ или АСТ, обострение подагры.
Побочные эффекты, отмеченные в постмаркетинговых исследованиях при терапии аторвастатином: анафилаксия, ангионевротический отек, буллезная сыпь (в т.ч.многоформная экссудативная эритема, синдром Стивенса-Джонсона, токсический эпидермальный некролиз), рабдомиолиз, разрыв сухожилия.
Взаимодействие
Риск развития миопатии при лечении статинами увеличивается при их использовании в сочетании с циклоспорином, фибратами, эритромицином, никотиновой кислотой в липидоснижающих дозах, сильными ингибиторами CYP3A4 (в т.ч. кларитромицином, ингибиторами ВИЧ-протеазы, итраконазолом).
При использовании аторвастатина в сочетании с ингибиторами изофермента CYP3A4(например циклоспорином, макролидными антибиотиками — эритромицином и кларитромицином; итраконазолом, ингибиторами ВИЧ-протеазы) могут повышаться плазменные концентрации аторвастатина (метаболизм аторвастатина осуществляется при участии CYP3A4); следует проявлять особую осторожность при использовании аторвастатина в сочетании с вышеупомянутыми средствами (см. «Особые указания»).
При комбинированном применении аторвастатина в дозе 40 мг и итраконазола в дозе 200 мг 1 раз в день было выявлено увеличение AUC аторвастатина в 3 раза по сравнению с AUCпри монотерапии.
Одновременное применение аторвастатина с ингибиторами протеаз (лопинавир/ритонавир, ритонавир/саквинавир) сопровождалось увеличением концентрации аторвастатина в плазме крови. При одновременном применении аторвастатина в дозе 40 мг и комбинации лопинавир+ритонавир (в дозе 400/100 мг 2 раза в день) было выявлено увеличение AUC аторвастатина в 5,9 раза по сравнению с AUC при монотерапии. При применении аторвастатина в дозе 40 мг с комбинацией ритонавир+саквинавир (в дозе 400/400 мг 2 раза в день) его AUC увеличивалось в 3,9 раза.
Антациды снижают концентрацию аторвастатина на 35% (влияние на содержание холестерина ЛПНП не меняется).
При повторном приеме дигоксина и аторвастатина Css дигоксина повышается примерно на 20% (за больными необходимо наблюдение).
При совместном применении аторвастатина и пероральных контрацептивов AUCнорэтиндрона и этинилэстрадиола увеличивается примерно на 30 и 20% (этот эффект следует учитывать при выборе перорального контрацептива для женщины, получающей аторвастатин).
При одновременном приеме с эритромицином (ингибитор CYP3A4) плазменная концентрация аторвастатина увеличивается примерно на 40%. Гиполипидемический эффект комбинации аторвастатина с колестиполом превосходит таковой для каждого препарата в отдельности. Одновременное применение с ЛС, снижающими концентрацию или активность эндогенных стероидных гормонов (в т.ч. кетоконазол, спиронолактон, циметидин), увеличивает риск снижения выработки эндогенных стероидных гормонов (следует соблюдать осторожность).
При одновременном приеме внутрь аторвастатина и суспензии, содержащей магния и алюминия гидроксид, концентрации аторвастатина в плазме снижались примерно на 35%, однако степень уменьшения уровня холестерина/ЛПНП при этом не менялась.
Передозировка
Лечение: симптоматическая и поддерживающая терапия. Специфического антидота нет. Гемодиализ неэффективен.
Пути введения
Внутрь.
Меры предосторожности вещества Аторвастатин
Перед началом и весь период лечения необходимо придерживаться стандартной гипохолестеринемической диеты.
Нарушение функции печени. Применение ингибиторов ГМГ-КоА-редуктазы для снижения уровня липидов в крови может приводить к изменению биохимических показателей, отражающих функцию печени. При проведении клинических испытаний у пациентов, получавших аторвастатин, частота стойкого повышения уровня трансаминаз в сыворотке (более чем в 3 раза выше уровня верхнего предела нормы, наблюдающееся в 2 или более случаях) составляла 0,7%. Частота этих нарушений при дозах 10, 20, 40 и 80 мг была 0,2; 0,2; 0,6 и 2,3%. У одного пациента развилась желтуха. Функцию печени следует контролировать перед началом лечения, через 6 нед, 12 нед после начала приема препарата и после каждого повышения дозы, а также периодически, например каждые 6 мес. Изменение активности ферментов печени обычно наблюдается в течение первых 3 мес после начала терапии. Пациенты, у которых отмечается повышение уровня трансаминаз, должны находиться под контролем до возвращения уровня ферментов к норме. В том случае, если значения АЛТ или АСТ более чем в 3 раза превышают уровень верхнего допустимого предела, рекомендуется снизить дозу или прекратить лечение.
Влияние на мышцы. В ряде случаев на фоне лечения аторвастатином у пациентов отмечалась миалгия, не приводившая к осложнениям. Пациенты с диффузной миалгией, вялостью или слабостью мышц и/или значительным повышением креатинфосфокиназы представляют собой группу риска в отношении развития миопатии (определяемой как боль в мышцах с сопутствующим повышением уровня креатинфосфокиназы более чем в 10 раз по сравнению с верхней границей нормы). При развитии миопатии (или предположении о ее наличии) необходимо определять активность креатинфосфокиназы; если значительное повышение ее уровня сохраняется, то рекомендуется снизить дозу или отменить аторвастатин.
При назначении сочетанной терапии аторвастатина с циклоспорином, производными фиброевой кислоты, эритромицином, кларитромицином, иммунодепрессантами и противогрибковыми препаратами азоловой структуры, а также никотиновой кислотой в дозах, вызывающих снижение уровня липидов, необходимо сопоставлять потенциальную пользу и степень риска и осуществлять наблюдение за пациентами, у которых появляются признаки или симптомы мышечных болей, вялости или слабости, особенно в течение первых месяцев лечения и при повышении дозы какого-либо из препаратов.
Препарат должен быть немедленно отменен при появлении признаков и наличии факторов риска развития острой почечной недостаточности вследствие рабдомиолиза (например острой тяжелой инфекции, артериальной гипотензии, обширного хирургического вмешательства, травмы, тяжелых метаболических и эндокринных нарушений, а также нарушений электролитного баланса).
Необходимо немедленно обратиться к врачу при появлении необъяснимых болей или слабости в мышцах, особенно если они сопровождаются недомоганием и лихорадкой.
Взаимодействия с другими действующими веществами
ПерейтиСвязанные новости
- Взаимодействие аторвастатина с другими лекарственными средствами
- Лекарственное взаимодействие статинов и ингибиторов протеазы
Торговые названия
Название | Значение Индекса Вышковского ® |
| Аторис® | 0,0497 |
| Липримар® | 0,047 |
| Торвакард® | 0,0372 |
| Тулип® | 0,016 |
| Аторвастатин | 0,0156 |
| Аторвастатин-Тева | 0,0061 |
| Новостат | 0,0023 |
| Атомакс® | 0,0017 |
| Аторвастатин-К | 0,0015 |
| Аторвастатин Алкалоид | 0,001 |
| Торвас | 0,0008 |
| Вазатор | 0,0007 |
| Атокорд | 0,0006 |
| Аторвастатин-СЗ | 0,0006 |
| Аторвастатин кальция | 0,0005 |
| Аторвастатина кальция тригидрат | 0,0005 |
| Аторвастатин-ЛЕКСВМ® | 0,0005 |
| Аторвокс | 0,0003 |
| Липофорд | 0,0003 |
| Аторвастатин кальция кристаллический | 0,0003 |
| Аторвастатин-OBL | 0,0003 |
| Атор | 0,0003 |
| Анвистат® | 0,0002 |
| АТОРВАСТАТИН-НАНОЛЕК | 0,0002 |
| ТГ-тор | 0,0001 |
| Липона | 0,0001 |
| Торвалип | 0,0001 |
| Аторвастатин МС | 0,0001 |
| АТОРВАСТАТИН АВЕКСИМА | 0 |
3. Ингибиторы микросомального триглицерид-переносящего белка
Аполипопротеин В (апоВ) играет ключевую роль в метаболизме липопротеинов. Ген апоВ человека находится на хромосоме 2 и продуцирует с помощью уникального процесса редактирования матричной рибонуклеиновой кислоты (мРНК) 2 физиологические изоформы циркулирующих липопротеинов – апоВ-48, имеющий м. м. 241 кДа и апоВ-100 с м. м. 512 кДа (5). Цифровые обозначения при букве «В» являются условными. Так, максимальная молекулярная масса апоВ принята за 100, а апоВ-48 имеет молекулярную массу, составляющую 48% от максимальной. Сейчас известно до 10 различных т. н. укороченных форм апопротеина В.
ApoB-48 (укороченная форма апоВ-100) продуцируется в кишечнике и играет важную роль при образовании, секреции хиломикронов. Синтез ApoB-100 происходит в печени. Этот белок является фундаментальным структурным компонентом липопротеинов очень низкой плотности (ЛПОНП) и продуктов их метаболизма – липопротеинов промежуточной плотности (ЛППП) и ЛПНП, а также лигандом для р-ЛПНП. Повышение в плазме концентрации апоВ свидетельствует об увеличении количества циркулирующих атерогенных липопротеинов.
Микросомальный триглицерид-переносящий белок – МТТР (Мicrosomal Тriglyceride Тransfer Рrotein) имеет важное значение для сборки и секреции апоВ-содержащих липопротеинов. МТТР впервые был описан в 1984 г. J. Wetterau и D. Zilversmit, выделившими данный белок из микросом печени быка (6). Он состоит из двух субъединиц (М и PDI). В состав М-субъединицы входит 894 аминокислоты (97 кДа). Эта субъединица экспрессируется прежде всего в гепатоцитах и энтероцитах.
Вторая субъединица – широко экспрессируемый многофункциональный белок дисульфид изомеразы – PDI (Protein Disulfide Isomerase) (55кДа) (7). Изомеразная активность PDI не является необходимым условием для функционирования МТТР. Хотя cубъединица PDI и не обладает активностью для переноса липидов, тем не менее она является важным звеном для поддержания активности всего комплекса в целом. Без PDI микросомальный триглицерид-переносящий белок образует нерастворимые агрегаты (8).
МТТР локализован вместе с PDI и ароВ в эндоплазматическом ретикулуме (ЭР) и аппарате Гольджи (АГ), стабилизируя образовавшийся полипептид апоВ и способствуя переносу нейтрального липида из мембраны ЭР в апоВ по принципу челночного механизма. Связывание МТТР с апоВ и перенос липида – независимые процессы. Активность МТР необходима для секреции апоВ-48 и апоВ-100.
3.1. МТТР в качестве терапевтической мишени
Исследования, которые проводились в 90-х годах прошлого века, установили, что ингибирование МТТР с использованием фармакологических препаратов приводит к снижению апоВ-содержащих липопротеинов, что дает возможность использовать данный процесс в лечении гиперлипидемии. Впервые в 1998 г. J. Wetterau и соавт. (9) описали небольшую молекулу ингибитора MTТP, она эффективно ингибировала секрецию апоВ-содержащих липопротеинов у грызунов и нормализовала гиперхолестеринемию у гиперлипидемических кроликов. Для этого использовали несколько синтетических ингибиторов MTТP (BMS-201038, СР-346086 и BAY-13-9953), эффективность которых была дозозависимой. Вместе с тем было отмечено, что у лабораторных животных, получавших данные соединения, наблюдалось обратимое накопление жира в печени и энтероцитах. Результаты экспериментов на животных позволяли прогнозировать развитие таких же побочных эффектов, которые могли возникнуть при клинических испытаниях ингибиторов МТТР на человеке. В связи с этим программа исследований была досрочно прекращена из-за развития таких нежелательных явлений, как нарушения со стороны желудочно-кишечного тракта и стеатоз печени (10). Однако спустя несколько лет работа по изучению ингибитора МТТР возобновилась, в результате чего появилось новое ЛС под названием «ломитапид».
3.2. Лекарственное средство ломитапид
Биотехнологическая компания Aegerion Pharmaceuticals, Inc. (США) сообщила об одобрении в декабре 2012 г. Управлением по контролю качества пищевых продуктов и лекарственных препаратов (FDA) препарата ломитапид (Lomitapide, Juxtapid). Показанием к применению данного ЛС является гомозиготная СГ – редкое наследственное заболевание, которое сопровождается очень ранним развитием атеросклероза.
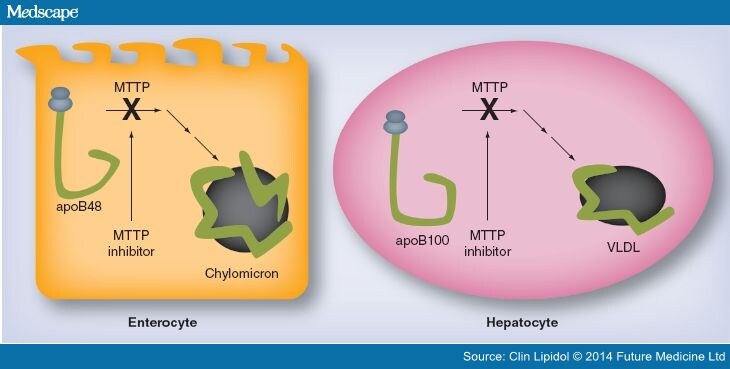
Ломитапид – мощный селективный ингибитор МТТР, внутриклеточного липид-переносящего белка, который обнаруживается в просвете эндоплазматического ретикулума и отвечает за связывание и транспортировку отдельных молекул липидов между мембранами. MTТP играет ключевую роль в сборке апоВ-содержащих липопротеинов в печени и кишечнике. Ингибирование МТР приводит к уменьшению синтеза ЛПОНП, ЛППП, ЛПНП и, как результат, – снижению уровня ЛПНП в крови (рис. 1).
![Lomitapide[mdash]mode of action.](http://www.nature.com/nrcardio/journal/v12/n10/carousel/nrcardio.2015.92-f2.jpg)

Lomitapide inhibits the activity of MTTP (1), thus inhibiting the loading of triglycerides onto ApoB and formation of VLDL particles. Consequently, secretion of VLDL from hepatocytes (2) is reduced, resulting in a reduction in LDL particles.
В одном из проводившихся ранее международных клинических исследований было показано, что пациенты с гомозиготной СГ, получавшие ингибитор МТТР, имели значительное снижение липидных показателей по сравнению с исходным уровнем: общий холестерин – ↓58,4%; ЛПНП – ↓50,9%; ЛПОНП – ↓78,7%; триглицериды – ↓65,4%.
Рекомендуемая начальная доза составляет 5 мг 1 раз в день. После 2 нед доза, с учетом приемлемой безопасности и переносимости, может быть увеличена до 10, а затем до 20 и 40 мг. Максимальная рекомендуемая доза – 60 мг.
4. Антисмысловые олигонуклеотиды
Антисмысловые олигонуклеотиды (АСО) представляют собой синтетические одноцепочечные молекулы дезоксирибонуклеиновой кислоты (ДНК) длиной от 8 до 50 нуклеотидов, которые целиком или частично связываются с рибонуклеиновой кислотой (РНК) и препятствуют дальнейшей трансляции мРНК в белок (11). Подавление биосинтеза белка под действием АСО происходит вследствие того, что мРНК-мишень в составе гибридного ДНК/РНК комплекса с АСО расщепляется внутриклеточной РНКазой Н (12) (рис. 2).
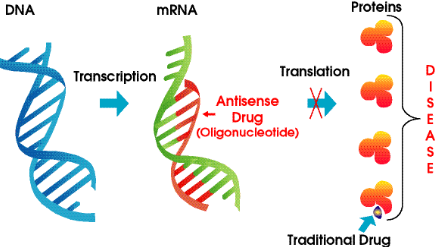
Впервые возможность избирательного подавления продукции определенного белка под действием АСО была продемонстрирована еще в 1978 г. в работе Р. Zamecnik и М. Stephenson (13). Авторы показали, что 13-звенный олигонуклеотид, комплементарный 3′-концевой последовательности РНК вируса саркомы Рауса, ингибирует репликацию вируса in vitro. Эта работа послужила толчком к изучению потенциала АСО в качестве ЛС для лечения вирусных заболеваний, воспалительных процессов, болезней крови, нарушений сердечно-сосудистой системы (14-18).
Ввиду того что природные олигодезоксирибонуклеотиды in vivo подвергаются быстрой деградации под действием нуклеаз, для повышения их стабильности в структуру АСО вносят различные химические модификации (19). Внесение модификаций в структуру олигонуклеотида не только повышает устойчивость АСО к нуклеазам, но и увеличивает эффективность их биологического действия, улучшает гибридизационные свойства и облегчает их захват клетками.
В первом поколении устойчивость к нуклеазам обеспечивалась модификацией фосфодиэфирной связи – замещение кислорода серой (фосфотиоаты), что позволяло повысить устойчивость препаратов к эндонуклеазам, не ухудшая при этом субстратные свойства комплекса относительно нуклеазы Н, однако затруднялось связывание АСО с целевой мРНК. Такие препараты АСО плохо проникали в клетку и имели ограниченный спектр распределения в органах и тканях. Препараты второго поколения были модифицированы по 2′-положению рибозы путем введения 2′-О-метильной, 2′-О-метоксиэтильной группы. Такая модификация позволяла повысить устойчивость к эндонуклеазам и снизить токсичность препарата, но ухудшала субстратные свойства дуплекса с целевой мРНК относительно РНКазы Н. Комбинация двух модификаций позволила получить АСО, обладающие хорошей специфичностью, селективностью и аффинностью к целевой молекуле (20). Химерные АСО, использующие одновременно фосфотиоатные и 2′-О-метоксиэтил или 2′-О-метил-модификации, легли в основу ЛС под названием «мипомерсен».
4.1. Лекарственное средство мипомерсен
Мипомерсен (Mipomerse Sodium, Kynamro) производства компании Genzyme Corporation, Isis Pharmaceuticals (США) одобрен FDA в январе 2013 г. Представляет собой синтетический 20-мерный 2′-О-(2-метокси)этил-модифицированный олигонуклеотид, второе поколение одноцепочечного АСО с конкретным комплементарным связыванием кодирующей области мРНК [нуклеотида 3249-3269] В-100 человека. Этот ингибитор имеет 20 нуклеотидов с кодирующей последовательностью 5′-GCCTCAGTCTGCTTCGCACC-3′ и является химически стабилизированным через фосфотиоат (21).
В нуклеотидную область мипомерсена включены сахара 2′-дезоксирибозы: 2′-O-метил (2’OMe) и 2′-O-метоксиметил (2’MOE) модифицированной рибозы в 3′ и 5′ концевых участках. В этом заключается отличие второго поколения АСО от первого, которое легко разрушается эндонуклеазой и экзонуклеазой и оказывает токсическое воздействие на рост и пролиферацию клеток здоровой ткани. Второе поколение АСО мипомерсена значительно эффективнее по сравнению с первым, поскольку обладает большей активностью, имеет повышенную устойчивость к деградации под действием ферментов нуклеаз, более длительный период полураспада и вызывает меньше побочных реакций (27-30).
Aнтисмысловые олигонуклеотиды предназначены для специфического связывания с мРНК апоВ-100 и предотвращения транслокации мРНК для формирования функционального апо-B100 (22-24). Эндогенные РНКазы ферментов, такие как РНКаза Н или Arganaute 2, распознают двойную мРНК апоB-100 и мипоперсена, разрушая тем самым цепь мРНК, в результате чего функциональный белок апоВ-100 не образуется (25,26). К тому же, разрушаясь под действием ферментов, апоВ-100 не экспрессирует из-за неправильного сплайсинга пре-мРНК для зрелой и функциональной мРНК.

Механизм действия мипомерсена (рис.) (31):
А – мипомерсен (одноцепочечный антисмысловой олигонуклеотид);
В – образуется антисмысловая двойная мРНК. После того как мипомерсен достигает гепатоцита, он проникает через мембрану клетки в ядро (механизм не ясен) и связывается с мРНК апоВ с высокой степенью точности;
С – РНКаза Н (неспецифическая эндонуклеаза, которая гидролизует фосфодиэфирные связи РНК при гибридизации с ДНК) расщепляет мРНК. После гибридизации с мРНК-мишенью мипомерсен-мРНК распознается и расщепляется эндогенной РНКазой Н, ферментом, участвующим в репликации/восстановлении ДНК;
D – результатом расщепления апoB мРНК является снижение синтеза апоВ и уменьшение ЛПНП в сыворотке.
В клинических исследованиях было продемонстрировано, что у пациентов, принимающих мипомерсен, среднее снижение липидных параметров (в %) от исходного уровня оставалось неизменным через 104 нед: ЛПНП – ↓27-28%; апоВ – ↓28-31%; липопротеин – ↓17-21%; ЛПВП – ↑3-10%; триглицериды – ↓3-14%.
Мипомерсен в дозе 200 мг вводится подкожно 1 раз в неделю.
![Mipomersen[mdash]mode of action.](http://www.nature.com/nrcardio/journal/v12/n10/images/nrcardio.2015.92-f1.jpg)
Антисмысловой олигонуклеотид связывается с мРНК ApoB и ингибирует трансляцию и синтез ApoB (1). В результате секреция частиц VLDL из гепатоцитов (2) снижается, что уменьшает образование частиц ЛПНП (3) и, как следствие, снижает уровень LDL-C в крови. Сокращения: ApoB, аполипопротеин B-100; CoA, кофермент A; HMG, 3-гидрокси-3-метилглютарил; LDL-C, LDL-холестерин; LPL, липопротеиновая липаза; MTTP, белок микросомального триглицеридного переноса; VLDL, липопротеин с очень низкой плотностью
5. Механизм регуляции PCSK9 экспрессии рецепторов ЛПНП на гепатоцитах
Печень является основным органом, отвечающим за клиренс
и катаболизм сывороточных ЛПНП. Гепатоциты регулируют уровень ЛПНП,
экспрессируя р-ЛПНП, которые связывают ЛПНП и удаляют их из плазмы.
Комплекс ЛПНП/р-ЛПНП интернализуется в гепатоцит в составе
клатриновых пузырьков, которые затем сливаются с эндосомами. Кислая среда
внутри эндосом способствует диссоциации комплекса ЛПНП/р-ЛПНП. После
диссоциации свободные р-ЛПНП повторно возвращаются на поверхность
гепатоцита, где они связывают и выводят из кровотока новые частицы
ЛПНП. Было показано, что на поверхности гепатоцита происходит
кальций-зависимое взаимодействие между каталитическим доменом PCSK9
и повтором А EGFР домена рецептора ЛПНП. После того как ЛПНП
связывается с таким рецептором, весь
комплекс – ЛПНП/р-ЛПНП/PCSK9 перемещается в клетку
в составе клатринового пузырька. В дальнейшем кислая среда эндосомы
способствует, с одной стороны,отделению ЛПНП от комплекса,
а с другой – повышению аффинности взаимодействия PCSK9
с р-ЛПНП за счет формирования дополнительных ионных связей между
про-доменом PCSK9 и р-ЛПНП. В результате PCSK9, не проявляя
протеолитической активности, удерживает, как распорка, р-ЛПНП в открытом
положении, не давая ему принять закрытую конформацию, необходимую для
повторного возвращения на поверхность клетки. Это ведет к тому, что
количество экспрессированных на гепатоците рецепторов снижается,
соответственно – уменьшается клиренс ЛПНП из сыворотки (35).
![PCSK9 inhibitors[mdash]mode of action.](http://www.nature.com/nrcardio/journal/v12/n10/images/nrcardio.2015.92-f3.jpg)
Моноклональные антитела нейтрализуют секретируемый PCSK9, что делает его неспособным связываться с LDLR (1). LDL связывается с LDLR в отсутствие PCSK9 (2), а LDL-LDLR-комплекс подвергается эндоцитозу (3). Как только в эндосоме комплекс LDL-LDLR диссоциирует, и LDL подвергается лизосомальной деградации, тогда как LDLR высвобождается (4); в результате, антитела PCSK9 повышают плотность LDLR на поверхности гепатоцитов (5), тем самым увеличивая поглощение частиц ЛПНП и снижая уровни LDL-C в крови. Сокращения: LDL-C, LDL-холестерин; LDLR, рецептор LDL; PCSK9, proprotein convertase subtilisin / kexin типа 9.

Существует несколько способов влияния на функцию PCSK9:
А – малые интерферирующие рибонуклеиновые кислоты (siRNAs) могут блокировать транскрипцию PCSK9 мессенджера рибонуклеиновой кислоты;
В – малые молекулярные ингибиторы могут нарушить процессинг PCSK9, что приведет к снижению образования функциональных PCSK9;
С, D – моноклональные антитела и аднектин ингибируют функцию PCSK9.
1 – SREBP-2 (стерол-связывающий регулирующий элемент-бета регулирует в гепатоцитах транскрипцию нескольких липидов и белков, в том числе рецепторов ЛПНП и PCSK9;
2 – PCSK9 превращается в эндоплазматическом ретикулуме в зрелую форму;
3 – упаковывается в аппарате Гольджи (как это делает рецептор ЛПНП) и затем секретируется;
4 – р-ЛПНП связывает циркулирующие ЛПНП;
5 – комплекс перемещается в эндосомы, где ЛПНП могут использоваться для других целей.
р-ЛПНП рециркулирует на клеточной поверхности до 150 раз, удаляя ЛПНП из циркуляции;
6 – PCSK9 регулирует количество р-ЛПНП путем их связывания и интернализации;
7 – разрушение происходит в лизосомах, что не позволяет рецепторам ЛПНП рециркулировать.
Указанные исследования дали понимание того, что снижение активности PCSK9 может стать терапевтической мишенью в лечении пациентов с гиперхолестеринемией, а это позволило бы изолированно снижать уровень ЛПНП. Блокада этого фермента приводит к экспрессии большего количества рецепторов ЛПНП на поверхности печени. В результате захват ЛПНП печенью увеличивается, их содержание в крови уменьшается. Блокирование PCSK9 является инновационным механизмом снижения ЛПНП. Из всех потенциальных ингибиторов PCSK9 для практического использования были выбраны моноклональные антитела человека (ЛС алирокумаб).
Что такое ингибиторы PCSK9?
Гиперлипидемия (повышенное содержание холестерина) - это хорошо известный фактор риска развития сердечно-сосудистых заболеваний. Текущие руководства для врачей подчеркивают необходимость лечения таких пациентов с использованием статинов в дозах, доказавших свою эффективность для снижения риска. Тем не менее, существуют ограничения для пациентов, которые либо не переносят терапию статинами, либо сохраняют гиперхолестеринемию на фоне максимально возможных доз этих лекарственных средств.
В 2003 г. был открыт ген, мутации которого могли приводить как к семейной гиперхолестринемии с ранним возникновением сердечно-сосудистых заболеваний, так и к сниженному содержанию атерогенных липопротеидов. Генетическая программа реализовывалась через уменьшение или увеличение активности пропротеиновой конвертазы субтилизин-кексинового типа 9 (PCSK9), печеночного фермента, играющего важную роль в обмене холестерина. Это открытие обусловило высокий интерес к разработке способов воздействия на PCSK9. Были использованы различные подходы к блокированию PCSK9, наиболее эффективным оказалось применение моноклональных антител. В настоящее время разработаны или разрабатываются по меньшей мере шесть моноклональных антител алирокумаб (ранее называемый SAR236553 / REGN727), эволокумаб (ранее называвшийся AMG145), RG7 652, LGT209 (NCT01979601, NCT01859455), 1B20 и бококизумаб (ранее называвшийся RN316 / PF-049 50615).
В 2015 г. Управление по контролю за продуктами и лекарствами США (FDA) одобрило два новых лекарственных средства для снижения уровня липопротеинов низкой плотности (ЛПНП) у пациентов с наследственной гиперхолестеринемией или пациентов с сердечно-сосудистыми заболеваниями, демонстрирующих высокие уровни ЛПНП, несмотря на соблюдение диеты и назначение максимально возможных доз статинов: эволокумаб (Репата, Repatha) и алирокумаб (Пралуент, Praluent) из класса ингибиторов PCSK9. Под воздействием ингибиторов PCSK9 уровни ЛПНП могут быть снижены на 50-60% ниже, чем при терапии статинами.
Особенности применения ингибиторов PCSK9
Алирокумаб (Пралуент)
Применяется путем подкожных инъекций и достигает максимальных концентраций независимо от места введения (живот, руки или бедра). Стабильная концентрация в крови достигается после введения 3 - 4 доз (75 – 150 мг каждая). Интервал между уколами – 2 недели. Максимальное снижение ЛПНП регистрируется в срок от 3 до 15 дней. Легкое или среднее нарушение функции почек или печени не препятствует использованию лекарства и не требует изменения дозы. Данные об использовании алирокумаба у пациентов с тяжелой печеночной или почечной недостаточностью отсутствуют.

Эволокумаб (Репата)
Препарат так же вводится в подкожную жировую клетчатку живота, бедра или верхние конечности при помощи одноразового инфузора с картриджем или нескольких последовательных инъекций автоинъектором или одноразовым шприцом. Препарат в дозе 140 мг вводится один раз в две недели, а в дозе 420 мг – один раз в месяц. Время достижения максимальной концентрации составляет от 3 до 4 дней после однократного применения. Максимальное снижение ЛПНП развивается при двухнедельном или одномесячном режиме введения через 14 дней.

Побочные эффекты и противопоказания
Следующие побочные эффекты были зарегистрированы при наблюдении за более чем 7000 пациентов (2476 для алирокумаба и 5416 для эволокумаба). Регистрировались все патологические состояния, достоверно отличающиеся по частоте от группы плацебо. Связь их с приемом препарата или ее отсутствие требуют специальных исследований.
Алирокумаб (Пралуент)
Препарат противопоказан пациентам, у которых возникли серьезные проявления гиперчувствительности, такие как васкулит или аллергические реакции, требующие госпитализации.
Наиболее частыми побочными эффектами, наблюдаемыми при применении алирокумаба, являются:
- назофарингит (11,3%),
- реакции на месте инъекции (покраснение кожи, зуд, отек, боль) (7,2%),
- грипп (5,7%),
- инфекция мочевых путей (4,8%),
- диарея (4,7%),
- бронхит (4,3%),
- мышечные боли (4,2%),
- мышечные судороги (3,1%),
- синусит (3%),
- кашель (2,5%),
- суставная и костная боль (2,1 %).
Наиболее частыми побочными эффектами, приводящими к прекращению приема лекарств, были аллергические реакции (0,6%) и повышение печеночных показателей (АЛТ) печени (0,3%).
Эволокумаб (Репата)
Противопоказания к применению эволокумаба сходны с таковыми для алирокумаба. Наиболее частыми побочными эффектами были:
- назофарингит (5,9%),
- инфекция верхних дыхательных путей (3,2%),
- боли в спине (3%),
- тошнота (2,1%).
Следует отметить, что наиболее распространенными побочными явлениями, приводящими к прекращению употребления препарата, были боль в мышцах, тошнота и головокружение. Среди других серьезных нежелательных явлений были сердечные расстройства у 2,4% пациентов, включая сердцебиение (0,6%), стенокардию (0,3%) и желудочковые экстрасистолы (0,3%).
Кроме того, данные исследований, оценивающих эволокумаб и алирокумаб, показали более высокую частоту когнитивных побочных эффектов у пациентов, получавших ингибиторы PCSK9 по сравнению со стандартной терапией. Однако они носили субъективный характер и не подтверждались тестированием. В настоящее время проводится специальное исследование, посвященное этому виду побочных эффектов (EBBINGHAUS).
Клиническое использование ингибиторов PCSK9
Считается, что использование ингибиторов PCSK9 станет особенно полезно при лечении больных с наследственной гиперхолестеринемией и тех, у кого невозможно достигнуть необходимых величин ЛПНП при использовании максимально приемлемых доз статинов.
Целевые уровни ЛПНП, по мере накопления исследовательских данных, постепенно снижаются. При этом достигать новых рубежей становится довольно сложно в связи с необходимостью назначения для этого максимальных доз липидоснижающих препаратов, что ведет к побочным эффектам. Предполагается, что ингибиторы PCSK9 могут быть использованы в комбинации со статинами, позволяя использовать их в небольших дозах, но при этом достигать целевых значений ЛПНП.
Существует несколько потенциальных барьеров, которые могут препятствовать широкому использованию ингибиторов PCSK9.
Во-первых, в настоящее время отсутствуют данные, демонстрирующие уменьшение частоты сердечно-сосудистых заболеваний при их применении (имеются данные только о воздействии на липидный профиль) и понадобится еще некоторое время для получения необходимой информации. Исследования, оценивающие влияние ингибиторов PCSK9 на долгосрочный прогноз сердечно-сосудистой заболеваемости сейчас проводятся (FOURIER, ODYSSEY)
Во-вторых, другим потенциальным барьером для широкого использования ингибиторов PCSK9 является их стоимость. В настоящее время годовые затраты на лечение оцениваются в $14350. Провести расчеты для России не представляется возможным, поскольку препараты официально не продаются в нашей стране. На черном рынке Пралуент предлагается по цене 770 Евро за дозу, что существенно превышает годовую бюджет, рассчитанный для США.
5.1. Моноклональные антитела, блокирующие фермент пропротеиновую конвертазу субтилизин-кексинового типа 9
В середине 80-х годов ХХ в. американские ученые J. Goldstein и М. Brown получили Нобелевскую премию по медицине и физиологии за открытие рецепторов ЛПНП и установление причин развития СГ. Результаты их исследований убедительно продемонстрировали, что ЛПНП удаляются из кровотока через рецептор-опосредованный путь и что экспрессия новых р-ЛПНП регулируется внутриклеточным содержанием холестерина по механизму отрицательной обратной связи (32). Однако механизмы разрушения уже существующих р-ЛПНП долгое время оставались непонятными. Только в 2003 г. был обнаружен ответственный за этот процесс фермент – пропротеин конвертаза, относящаяся к семейству сериновых протеаз и получившая название «пропротеиновая конвертаза субтилизин-кексинового типа 9» (PCSK9 – от Proprotein Convertase Subtilisin/Kexin type 9). PCSK9 – фермент-гидролаза, продукт гена человека PCSK9. Относится к семейству пропротеиновых конвертаз, которые активируют ферменты, отщепляя от них пептид, ингибирующий их каталитическую активность. PCSK9 играет важную роль в гомеостазе холестерина и является потенциальной мишенью для агентов, снижающих уровень холестерина ЛПНП в крови. Показано, что синтезированная в печени PCSK9 секретируется в кровоток и образует комплекс с р-ЛПНП на поверхности гепатоцита. Не проявляя протеолитической активности, PCSK9 способствует разрушению рецептора после интернализации образовавшегося комплекса в клетку (33,34).
5.2. Лекарственное средство алирокумаб
Алирокумаб (Alirocumab, Praluent), совместная разработка фармацевтической компании Sanofi (Франция) и биофармацевтической компании Regeneron Pharmaceuticals (США), одобрен FDA в октябре 2015 г. Препарат представляет собой моноклональные антитела человека (изотип IgG1), которые связываются с PCSK9 с высокой аффинностью и специфичностью и производятся с использованием технологии рекомбинантной ДНК в суспензионной культуре клеток яичников китайского хомячка. Алирокумаб состоит из двух дисульфид-связанных тяжелых цепей, каждая из которых ковалентно связана с легкими каппа-цепями. Единственный N-связанный сайт гликозилирования локализован в каждой тяжелой цепи на CH2 домене Fc константной области молекулы. Вариабельные домены тяжелой и легкой цепей в совокупности образуют сайт связывания PCSK9. Приблизительная молекулярная масса алирокумаба составляет 146 кДа.
Механизм действия алирокумаба показан на рисунке

Рекомендуемая начальная доза алирокумаба составляет 75 мг. Вводят подкожно 1 раз в 2 нед. Если ответ ЛПНП является неадекватным, доза может быть увеличена до 150 мг.
Появление на мировом фармацевтическом рынке новых гиполипидемических препаратов с уникальным механизмом действия, удобным режимом дозирования и высокой эффективностью предоставляет врачам-клиницистам более широкие возможности в выборе средств терапии пациентов с дислипидемией.